Ответ на повреждение атеросклероз
Клиническое значение атеросклероза и его осложнений пробудило огромный интерес к исследованию механизмов, лежащих в основе этого заболевания. Ранее были выдвинуты две основные гипотезы: одна из них подчеркивает значение пролиферации клеток интимы, другая фокусирует внимание на рецидивирующем тромбообразовании и организации тромбов.
Современные представления об атерогенезе заимствуют элементы обеих теорий и добавляют описанные ранее факторы риска. Согласно гипотезе «ответ на повреждение» атеросклероз представляет собой хроническую воспалительную реакцию и процесс заживления артериальной стенки в ответ на повреждение эндотелия. Поражение прогрессирует в результате взаимодействия модифицированных липопротеинов, макрофагов моноритарного происхождения и Т-лимфоцитов с нормальными клеточными компонентами артериальной стенки.
Соответственно этой модели атеросклероз обусловливают следующие патогенетические процессы:
— хроническое повреждение эндотелия приводит к повышенной сосудистой проницаемости, адгезии лейкоцитов и тромбозу;
— накопление липопротеинов (в основном ЛПНП и их окисленных форм) в стенке сосудов;
— адгезия моноцитов к эндотелию с их последующей миграцией и трансформацией в макрофаги и пенистые клетки;
— адгезия тромбоцитов;
— высвобождение факторов активированными тромбоцитами, макрофагами и клетками сосудистой стенки, включая миграцию гладкомышечных клеток либо из медии сосудистой стенки, либо из циркулирующих клеток-предшественников;
— пролиферация гладкомышечных клеток и образование ВКМ;
— накопление липидов как вне, так и внутри клеток (макрофагов и гладкомышечных клеток).
Теперь детально рассмотрим основные механизмы атерогенеза.
а) Повреждение эндотелия. В основе гипотезы «ответ на повреждение» лежит дисфункция эндотелия, а не утрата его вследствие любого типа повреждения (путем механического слущивания, под влиянием гемодинамических сил, отложения иммунных комплексов, облучения или химических веществ), когда происходит лишь утолщение интимы. Ранние повреждения стенки сосуда в случае богатой липидами диеты и других факторов риска появляются в участках морфологически интактного эндотелия, но с нарушенной функцией. Такой эндотелиальный слой имеет повышенную проницаемость, усиленную адгезию лейкоцитов и измененную экспрессию генов.
Специфические этиологические факторы, способствующие дисфункции эндотелия при раннем атеросклерозе, изучены недостаточно полно. К этим факторам относятся гипертензия, гиперлипидемия, токсичные вещества табачного дыма, гипергомоцистеинемия и инфекции. Воспалительные цитокины, в частности фактор некроза опухолей (TNF), также могут стимулировать проатерогенные формы экспрессии генов эндотелиальных клеток. Наиболее важные причины дисфункции эндотелия — гемодинамические нарушения и гиперхолестеринемия.
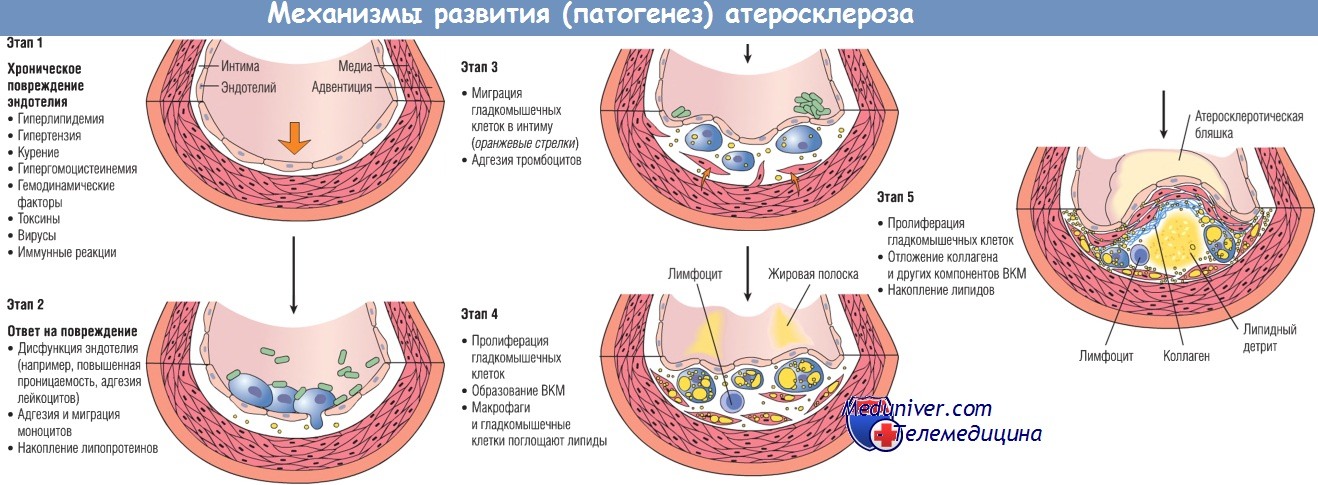
Развитие изменений артериальной стенки согласно гипотезе «ответ на повреждение».
ВКМ — внеклеточный матрикс.
б) Гемодинамические нарушения. Значение гемодинамических нарушений в патогенезе атерогенеза иллюстрирует факт, что бляшки имеют тенденцию локализоваться в устьях артерий крупного калибра, участках ветвления артерий и вдоль задней стенки брюшной аорты, т.е. там, где кровоток имеет особые характеристики. Исследования in vitro показали, что нетурбулентный ламинарный кровоток в нормальной сосудистой сети индуцирует гены, продукты которых (например, антиоксидантная супероксиддисмутаза) защищают от атеросклероза. Такие «атеропротективные» гены объясняют неслучайную локализацию ранних атеросклеротических поражений.
в) Липиды. Липиды обычно транспортируются с кровотоком, будучи связанными со специфическими апопротеинами (в виде липопротеиновых комплексов). Дислипопротеинемия является результатом мутаций, изменяющих апопротеины или клеточные рецепторы липопротеинов, а также результатом других состояний, влияющих на уровень липидов (например, нефротического синдрома, алкоголизма, гипотиреоза, сахарного диабета). К частым аномалиям липидного статуса (присутствующим у многих пациентов, перенесших инфаркт миокарда) относятся:
(1) повышенный уровень ЛПНП;
(2) сниженный уровень ЛПВП;
(3) повышенный уровень аномального липопротеина (а).
Причастность гиперхолестеринемии к атерогенезу доказывают следующие данные:
— преобладающими липидами в атеросклеротических бляшках являются холестерин и его эфиры;
— генетические дефекты захвата и метаболизма липопротеинов, обусловливающие липопротеинемию, ассоциируются с ранним атеросклерозом. Так, гомозиготная семейная гиперлипопротеинемия из-за дефекта рецепторов ЛПНП и неадекватного поглощения ЛПНП в печени может вызвать инфаркт миокарда до возраста 20 лет. Ранний атеросклероз получен в экспериментах на животных с генетически созданным дефицитом аполипопротеинов или рецепторов ЛПНП;
— другие генетические или приобретенные заболевания (например, сахарный диабет, гипотиреоз), вызывающие гиперхолестеринемию, способны стать причиной раннего атеросклероза;
— эпидемиологический анализ показал корреляцию между тяжестью атеросклероза и уровнем общего холестерина или ЛПНП в плазме;
— снижение уровня холестерина в сыворотке при соответствующей диете или на фоне приема лекарственных средств замедляет скорость прогрессирования атеросклероза, вызывает регрессию некоторых бляшек и снижает риск сердечнососудистых осложнений.
Механизмы, посредством которых хроническая гиперлипидемия способствует атерогенезу:
— хроническая гиперлипидемия, особенно гиперхолестеринемия, может непосредственно нарушать функцию эндотелиальных клеток, повышая местную продукцию свободных радикалов кислорода, называемых активными формами кислорода (АФК). АФК способны повреждать ткани за счет ускоренного распада оксида азота, что снижает его вазодилататорную активность;
— при хронической гиперлипидемии липопротеины накапливаются в интиме. Эти липиды окисляются под действием АФК, продуцируемых макрофагами или эндотелиальными клетками местно. Окисленные ЛПНП поглощаются макрофагами с помощью скавенджер-рецепторов, отличающихся от рецепторов ЛПНП, и накапливаются в фагоцитах (пенистых клетках). Кроме того, окисленные ЛПНП стимулируют высвобождение факторов роста, цитокинов и хемоки-нов эндотелиальными клетками и макрофагами, что усиливает миграцию моноцитов в поврежденные участки. Наконец, окисленные ЛПНП цитотоксичны для эндотелиальных и гладкомышечных клеток и способны индуцировать дисфункцию эндотелия. На роль окисленных ЛПНП в атерогенезе указывает факт их накопления в макрофагах на всех стадиях формирования атеросклеротических бляшек.
г) Воспаление. Процесс воспаления способствует инициации, прогрессированию и развитию осложнений атеросклероза. В норме сосудистая стенка не адгезирует воспалительные клетки, но уже на ранних стадиях атерогенеза артериальные эндотелиальные клетки с нарушенной функцией экспрессируют молекулы, стимулирующие адгезию лейкоцитов. В частности, молекула адгезии сосудистого эндотелия 1 (VCAM-1) обеспечивает адгезию моноцитов и Т-клеток к эндотелию. После этого они мигрируют в интиму под влиянием местно продуцируемых хемокинов.
Моноциты трансформируются в макрофаги и с высокой авидностью поглощают липопротеины, включая ЛПНП. Миграция и дифференцировка моноцитов в макрофаги (и в конечном итоге в пенистые клетки) теоретически носит протективный характер, поскольку эти клетки удаляют потенциально болезнетворные липидные частицы. Однако окисленные ЛПНП стимулируют активацию макрофагов и продукцию цитокинов (например, TNF). Это еще больше повышает адгезию лейкоцитов и продукцию хемокинов (например, моноцитарного хемоаттрактантного белка 1), что дает стимул для миграции дополнительного количества мононуклеарных воспалительных клеток. Активированные макрофаги продуцируют также реактивные метаболиты кислорода, усиливающие окисление ЛПНП, и секретируют факторы роста, индуцирующие пролиферацию гладкомышечных клеток.
Т-лимфоциты, привлеченные в интиму, взаимодействуют с макрофагами и поддерживают хроническую воспалительную реакцию. Нет четкого представления, реагируют ли Т-клетки на специфические антигены (например, на бактериальные или вирусные антигены, на белки теплового шока, на модифицированные составляющие сосудистой стенки и липопротеины) или они неспецифически активируются в условиях локального воспаления. Как бы там ни было, активированные Т-клетки вырабатывают в зоне повреждения интимы воспалительные цитокины (например, IFN-y), которые могут стимулировать макрофаги, эндотелиальные и гладкомышечные клетки.
Вследствие хронического воспаления активированные лейкоциты и клетки сосудистой стенки высвобождают факторы роста, стимулирующие пролиферацию гладкомышечных клеток и синтез ВКМ.
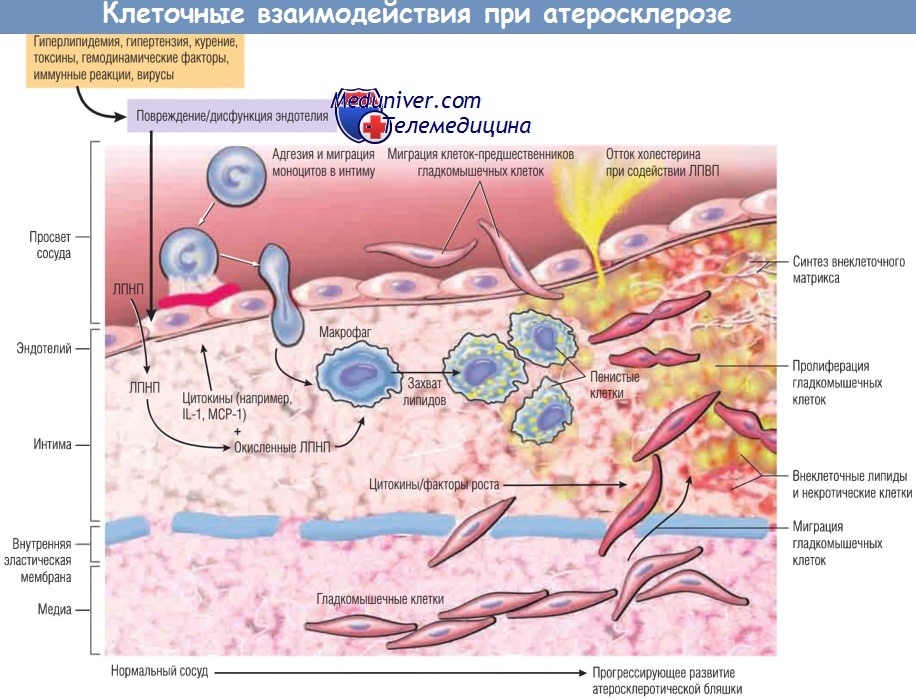
Гипотеза клеточных взаимодействий при атеросклерозе.
Предполагается, что гиперлипидемия и другие факторы риска повреждают эндотелий,
что приводит к адгезии тромбоцитов и моноцитов и высвобождению факторов роста (включая тромбоцитарный фактор роста),
индуцирующих миграцию и пролиферацию гладкомышечных клеток.
Пенистые клетки атеросклеротических бляшек образуются из макрофагов с помощью рецепторов липопротеинов очень низкой плотности и скавенджер-рецепторов модифицированных липопротеинов низкой плотности (ЛПНП),
например окисленных ЛПНП, и гладкомышечных клеток (механизм образования менее изучен).
Внеклеточные липиды попадают в бляшку из просвета сосуда, особенно в случае гиперхолестеринемии, а также из дегенерирующих пенистых клеток.
Накопление холестерина в бляшке отражает дисбаланс между притоком и оттоком холестерина,
а липопротеины высокой плотности (ЛПВП), вероятно, способствуют выведению холестерина из бляшки.
Гладкомышечные клетки мигрируют в интиму, пролиферируют и продуцируют ВКМ, включая коллаген и протеогликаны.
IL-1 — интерлейкин-1; МСР-1 — моноритарный хемоаттрактантный белок 1.
д) Инфекция. Было бы заманчиво предположить, что инфекция может индуцировать местный воспалительный процесс, лежащий в основе атеросклероза, однако эта гипотеза еще нуждается в доказательстве. В атеросклеротических бляшках обнаружены герпес-вирус, цитомегаловирус и С. pneumoniae, и у пациентов с тяжелым атеросклерозом сероэпидемиологические данные указывают на повышенный титр антител к С.pneumoniae. Не ясно, участвует ли С. pneumoniae в патогенезе атеросклероза, поскольку она часто ассоциируется с бронхитом и курением, которое является фактором риска ИБС. Данная инфекция широко распространена (как и атеросклероз), поэтому трудно определить, есть ли причинная связь или это просто совпадение.
Тем не менее вполне возможно, что микроорганизмы способны инфицировать атеросклеротические бляшки на ранней стадии их формирования, при этом чужеродные антигены могут потенцировать атерогенез, индуцируя местный иммунный ответ, или инфекционный агент способствует развитию местного протромботического статуса.
и) Пролиферация гладкомышечных клеток. Пролиферация гладкомышечных клеток и формирование ВКМ превращают жировые полоски (наиболее ранняя стадия поражения) в зрелую атеросклеротическую бляшку и способствуют ее увеличению. (Напомним, что гладкомышечные клетки интимы могут быть производными клеток-предшественников, циркулирующих в крови, и характеризуются пролиферативным и синтетическим фенотипами, что отличает эти клетки от гладкомышечных клеток подлежащей медии.) В пролиферации этих клеток и синтезе ВКМ участвуют некоторые факторы роста, включая PDGF (высвобождаемый локально прилипшими тромбоцитами, а также макрофагами, эндотелиальными и гладкомышечными клетками), фактор роста фибробластов и трансформирующий фактор роста a (TGF-a).
Мигрировавшие гладкомышечные клетки синтезируют ВКМ (в т.ч. коллаген), стабилизирующий атеросклеротические бляшки, но активированные воспалительные клетки в атеросклеротической бляшке могут вызвать апоптоз гладкомышечных клеток интимы, а также повысить катаболизм ВКМ, приводя к нестабильности бляшек.
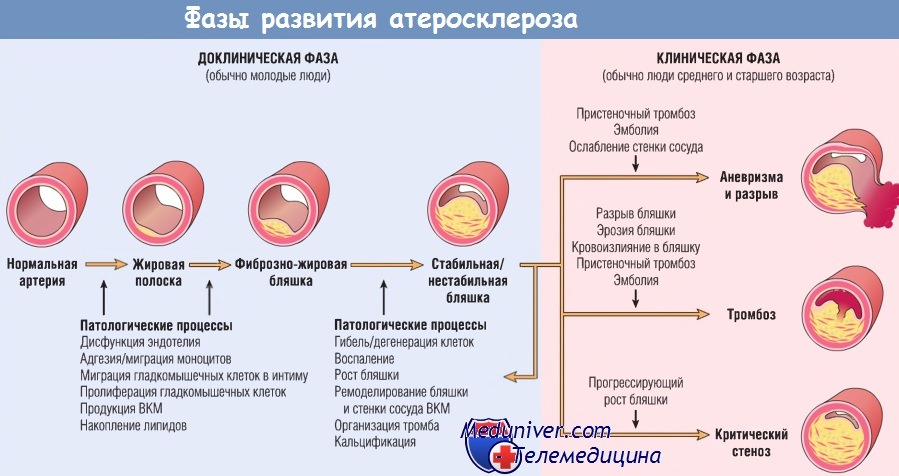
На рисунке ниже проиллюстрирована гипотеза развития атеросклероза как хронической воспалительной реакции (вместе с процессами, направленными в конечном итоге на попытку сосудистого «исцеления»), индуцированной разнообразными воздействиями, включая повреждение эндотелия, накопление и окисление липидов и тромбоз. Атеросклеротические бляшки — патологические динамические образования, состоящие из эндотелиальных клеток с нарушенной функцией, мигрировавших и пролиферирующих гладкомышечных клеток с примесью лимфоцитов и макрофагов. Все эти клетки способны секретировать медиаторы, влияющие на атерогенез.
Таким образом, бляшки в интиме на ранних стадиях развития немного больше по размеру, чем агрегаты, состоящие из гладкомышечных и макрофагальных пенистых клеток. По мере прогрессирования патологического процесса атеросклеротическая бляшка видоизменяется за счет ВКМ, синтезируемого гладкомышечными клетками. В интиме особенно хорошо выражена соединительная ткань, образующая фиброзную покрышку бляшки, при этом поражение обычно сохраняет центральное ядро из нагруженных липидами клеток и липидного детрита, который может кальцифицироваться. Со временем бляшка в интиме все больше выступает в просвет сосуда или сдавливает подлежащую медию, вызывая ее дегенерацию. Разрыв фиброзной покрышки может привести к тромбозу и острой окллюзии сосуда.
— Рекомендуем ознакомиться со следующей статьей «Морфология атеросклероза»
Оглавление темы «Атеросклероз и его последствия»:
- Причины и факторы риска атеросклероза
- Механизмы развития (патогенез) атеросклероза
- Морфология атеросклероза
- Последствия и осложнения атеросклероза
- Механизмы развития аневризмы и ее расслоения
- Механизмы развития и морфология аневризмы брюшной аорты
- Признаки аневризмы брюшной аорты
- Признаки аневризмы грудной аорты
- Механизмы развития (патогенез) расслоения аорты
- Морфология расслоения аорты
Несмотря на многообразие существующих теорий липогенеза, ключевая роль следующих 4-х факторов в патогенезе атеросклероза признается всеми. Это:
-эндотелиоциты;
-моноциты;
-гладкомышечные клетки артерии;
-гиперлипидемия.
В настоящее время наиболее популярна гипотеза “Ответ на повреждение”, в соответствии с которой атеросклероз рассматривается как реакция на повреждение эндотелия сосудистой стенки. Под повреждением подразумевается нарушение его обычных функций, которое проявляется повышением проницаемости и адгезивности, увеличением секреции прокоагулянтных и сосудосуживающих факторов. Эти изменения эндотелия происходят под действием вышеописанных факторов риска атеросклероза токсические соединения, например, компоненты табачного дыма, избыточный уровень гормонов, например, гиперинсулинемия при сахарном диабете II типа, гемодинамические факторы, например, артериальная гипертензия, даже доказана роль инфекционных агентов в повреждении эндотелия, в частности, вирусов герпеса.
Однако в качестве наиболее важного повреждающего фактора выступает гиперхолестеринемия. При гиперхолестеринемии в мембране эндотелиальных клеток увеличивается содержание ХС и соотношение ХС/фосфолипиды, что приводит к повышению ее проницаемости для ЛПНП. Однако, при пассаже через эндотелий ЛПНП подвергаются окислению, приобретая более агрессивные свойства, способствующие еще большему повреждению эндотелия и интимы. В результате возникает избыточная инфильтрация интимы ЛПНП.
Следующим этапом атерогенеза является инфильтрация интимы макрофагами, сфагоцитировавшими окисленные ЛПНП, которые разрушаются с образованием эфиров холестерина, в результате чего макрофаги превращаются в так называемые пенистые клетки. Образуются липидные полоски — первая морфологическая стадия атеросклеротической бляшки.
Макрофаги играют роль и в дальнейшем прогрессировании атеросклеротических изменений сосудов. Они секретируют биологически активные соединения, включая хемотаксины, митогены и факторы роста, которые стимулируют миграцию из медии в интиму гладкомышечных клеток (ГМК) и фибробластов, их пролиферацию, репликацию и синтез соединительной ткани.
Далее происходит миграция из медии в интиму ГМК и фибробластов с их последующей пролиферацией под влиянием стимуляторов (факторов роста), синтезируемых макрофагами, тромбоцитами, агрегированными в месте повреждения эндотелия, лейкоцитами. Пролиферация гладкомышечных клеток сопровождается синтезом матриксных белков – коллагена, эластических волокон и протеогликанов. Помимо синтеза соединительной ткани ГМК способны захватывать ЛПНП и трансформироваться в пенистые клетки, как и макрофаги
На ранних стадиях атеросклероз представлен липидными полосками, которые содержат пенистые клетки, богатые ХС и его эфирами. В последующем вокруг зоны накопления липидов развивается соединительная ткань и происходит формирование фиброзной атеросклеротической бляшки. Этот процесс обычно занимает несколько десятилетий.
Воспалительная гипотеза атеросклероза. Можно заметить, что в патогенезе атеросклероза происходят закономерности, свойственные любому воспалению: воздействие повреждающего фактора (окисленных ЛПНП), клеточная инфильтрация, фагоцитоз и формирование соединительной ткани (по аналогии со стадиями воспаления – альтерация, экссудация, пролиферация). Сходство с воспалением подтверждается тем, что на любых этапах формирования атеросклеротической бляшки присутствуют Т-лимфоциты, что свидетельствует о роли иммунных, а возможно, и аутоиммунных механизмов. Возможно, антигенами, на которые реагируют Т-лимфоциты, являются окисленные ЛПНП. Таким образом, атеросклероз рассматривается как воспаление в ответ на повреждение эндотелия факторами риска.
Гипотеза: “Ответ на удерживание частиц”. В экспериментах последних лет доказано, что взаимодействие между аполипопротеинами и рецепторами клеток эндотелия происходит раньше, чем повреждение эндотелия. При этом считается, что наиболее мелкие и атерогенные субфракции липопротеинов низкой плотности (ЛПНП), могут свободно проникать через эндотелиальный барьер и накапливаться в субэндотелиальном пространстве. При минимальной степени окисления частицы воздействуют на эндотелий, вызывая синтез адгезинов, моноцитарного колониестимулирующего фактора, тканевого фактора, моноцитарного хемоаттрактивного протеина, активатора ингибитора плазминогена. Более существенное окисление ЛПНП приводит к выраженной модификации частиц и интенсивному захвату их макрофагами с последующий миграцией их в интиму и превращением в пенистые клетки.
Моноклональная гипотезагласит, что атеросклероз – клональный неопластический процесс, который возникает в результате соматической мутации в гладкомышечных клетках артерий, по типу доброкачественной лейомиомы. В пользу этой гипотезы говорят факты о том, что во многих атеромах пенистые клетки являются потомством одной клетки-предшественницы.
Инфекционные гипотезы атерогенеза во главу угла ставят инфицирование некоторыми микроорганизмами, в частности, вирусом герпеса, хламидиями, чему находят иммунологическое и эпидемиологическое подтверждение.
Дата добавления: 2015-05-13; просмотров: 601; Опубликованный материал нарушает авторские права? | Защита персональных данных | ЗАКАЗАТЬ РАБОТУ
Не нашли то, что искали? Воспользуйтесь поиском:
Лучшие изречения: Увлечёшься девушкой-вырастут хвосты, займёшься учебой-вырастут рога 10152 — | 7889 — или читать все…
Читайте также:

