Анемия у детей талассемия

Талассемия – наследственные гемоглобинопатии, характеризующиеся угнетением синтеза цепочечных белковых молекул, образующих структуру гемоглобина. Это приводит к повреждению мембраны эритроцитов и разрушению красных клеток крови с развитием гемолитических кризов. Признаками талассемии служат характерные костные изменения, гепатоспленомегалия, анемический синдром. Диагноз талассемии подтверждается клиническими и лабораторными данными (исследованием гемограммы, гемоглобина, миелограммы, электрофоретическим методом). Возможна пренатальная диагностика талассемии. В лечении талассемии применяются гемотрансфузии, терапия десфералом, спленэктомия, трансплантация костного мозга.
Общие сведения
Талассемия – группа генетически детерминированных болезней крови, развивающихся при нарушении синтеза a- или β-цепей гемоглобина, сопровождающихся гемолизом, гипохромной анемией, микроцитозом. В гематологии талассемия относится к наследственным гемолитическим анемиям — количественным гемоглобинопатиям. Талассемия широко распространена среди населения Средиземноморского и Черноморского региона; название заболевания буквально переводится как «анемия морского побережья». Также случаи талассемии нередки в странах Африки, Ближнего Востока, Индии и Индонезии, Средней Азии и Закавказья. С синдромом талассемии каждый год в мире рождается 300 тыс. детей. В зависимости от формы патологии течение талассемии может быть тяжелым, фатальным или легким, бессимптомным. Так же, как серповидно-клеточная анемия, талассемия играет роль защитного фактора против малярии.

Талассемия
Классификация талассемии
С учетом поражения той или иной полипептидной цепи гемоглобина различают:
- a-талассемию (с подавлением синтеза альфа-цепей HbA). Данная форма может быть представлена гетерозиготным носительством манифестного (α-th1) или немого (α-th2) гена; гомозиготной a-талассемией (водянкой плода с гемоглобином Бартса); гемоглобинопатией Н
- b-талассемию (с подавлением синтеза бета-цепей HbA). Включает в себя гетерозиготную и гомозиготную β-талассемию (анемию Кули), гетерозиготную и гомозиготную δβ-талассемию (F-талассемию)
- γ-талассемию (с подавлением синтеза гамма-цепей гемоглобина)
- δ-талассемию (с подавлением синтеза дельта-цепей гемоглобина)
- талассемию, обусловленную нарушением структуры гемоглобина.
Статистически чаще встречается β-талассемия, которая, в свою очередь, может протекать в 3-х клинические формах: малой, большой и промежуточной. По тяжести синдрома выделяют легкую форму талассемии (пациенты доживают до половой зрелости), средне-тяжелую (продолжительность жизни больных 8-10 лет) и тяжелую (гибель детей наступает в первые 2-3 года жизни).
Причины талассемии
Талассемия является генетическим заболеванием с аутосомно-рецессивным наследованием. Непосредственной причиной патологии выступают различные мутационные нарушения в гене, кодирующем синтез той или иной цепи гемоглобина. Молекулярную основу дефекта могут составлять синтез аномальной матричной РНК, делеции структурных генов, мутации регуляторных генов либо их неэффективная транскрипция. Следствием подобных нарушений служит снижение или отсутствие синтеза одной из полипептидных гемоглобиновых цепей.
Так, при b-талассемии бета-цепи синтезируются в недостаточном количестве, что приводит к избытку альфа-цепей, и наоборот. Избыточно продуцируемые полипептидные цепи откладываются в клетках эритроидного ряда, вызывая их повреждение. Это сопровождается деструкцией эритрокариоцитов в костном мозге, гемолизом эритроцитов в периферической крови, гибелью ретикулоцитов в селезенке. Кроме этого, при b-талассемии в эритроцитах накапливается фетальный гемоглобин (НbF), не способный транспортировать кислород в ткани, что вызывает развитие тканевой гипоксии. Вследствие костномозговой гиперплазии развивается деформация костей скелета. Анемия, тканевая гипоксия и неэффективный эритропоэз в той или иной степени нарушают развитие и рост ребенка.
Для гомозиготной формы талассемии характерно наличие двух дефектных генов, унаследованных от обоих родителей. При гетерозиготном варианте талассемии пациент является носителем мутантного гена, унаследованного от одного из родителей.
Симптомы талассемии
Признаки большой (гомозиготной) b-талассемии проявляются уже в течение 1-2-го года жизни ребенка. Больные дети имеют характерное монголоидное лицо, седловидную переносицу, башенный (четырехугольный) череп, гипертрофию верхней челюсти, нарушение прикуса, гепато- и спленомегалию. Проявлениями анемизации служат бледный или землисто-желтушный цвет кожных покровов. Поражение трубчатых костей сопровождается отставанием в росте и патологическими переломами. Возможно развитие синовита крупных суставов, калькулезного холецистита, язв нижних конечностей. Фактором, осложняющим течение b-талассемии, выступает гемосидероз внутренних органов, приводящий к развитию цирроза печени, фиброза поджелудочной железы и, как следствие, — сахарного диабета; кардиосклероза и сердечной недостаточности. Больные восприимчивы к инфекционным заболеваниям (кишечным инфекциям, ОРВИ и др.), возможно развитие тяжелых форм пневмонии и сепсиса.
Малая (гетерозиготная) b-талассемия может протекать бессимптомно или с минимальными клиническими проявлениями (умеренным увеличением селезенки, незначительно выраженной гипохромной анемией, жалобами на повышенную утомляемость). Аналогичная симптоматика сопровождает течение гетерозиготной формы a-талассемии.
При гомозиготной форме a-талассемии альфа-цепи полностью отсутствуют; фетальный гемоглобин у плода не синтезируется. Данная форма талассемии несовместима с жизнью, что приводит к внутриутробной гибели плода вследствие развивающегося синдрома водянки или самопроизвольному прерыванию беременности. Течение гемоглобинопатии Н характеризуется развитием гемолитической анемии, спленомегалии, тяжелых костных изменений.
Диагностика талассемии
Талассемию следует заподозрить у лиц с семейным анамнезом, характерными клиническими признаками и лабораторными показателями. Больные талассемией нуждаются в консультации гематолога и медицинского генетика.
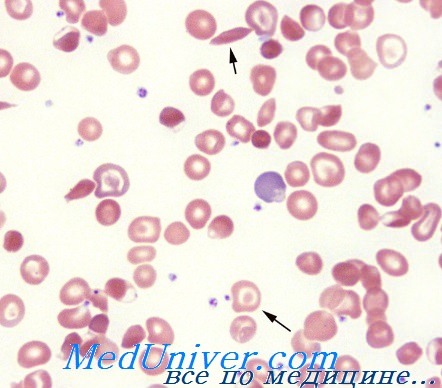
Типичными гематологическими изменениями служат снижение уровня гемоглобина и цветового показателя, гипохромия, наличие мишеневидных эритроцитов, повышение уровня железа сыворотки крови и непрямого билирубина. Электрофорез Hb на ацетат-целлюлозной пленке используется для определения различных гемоглобиновых фракций. При изучении пунктата костного мозга обращает внимание гиперплазия красного кроветворного ростка с высоким числом эритробластов и нормобластов. Молекулярно-генетические исследования позволяют выявить мутацию в локусе a- или β-глобина, нарушающую синтез полипептидной цепи.
На краниограммах при большой b-талассемии выявляется игольчатый периостоз (феномен «волосатого черепа»). Характерна поперечная исчерченность трубчатых и плоских костей, наличие мелких очагов остеопороза. С помощью УЗИ брюшной полости обнаруживается гепатоспленомегалия, камни желчного пузыря.
При подозрении на талассемию требуется исключить железодефицитную анемию, наследственный микросфероцитоз, серповидно-клеточную анемию, аутоиммунную гемолитическую анемию. В семьях, имеющих больных талассемией, рекомендуется проведение генетического консультирования супругов и инвазивной дородовой диагностики (биопсии хориона, кордоцентеза, амниоцентеза) для выявления гемоглобинопатии на ранних сроках беременности. Подтверждение гомозиготных форм талассемии у плода служит показанием для искусственного прерывания беременности.
Лечение и прогноз талассемии
Лечебная тактика при различных формах талассемии неодинакова. Так, пациенты с малой b-талассемией в лечении не нуждаются. С другой стороны, больным с гомозиготной b-талассемией с первых месяцев жизни требуется проведение гемотрансфузионной терапии (переливание размороженных или отмытых эритроцитов), введение хелатирующих препаратов, связывающих железо (дефероксамина), глюкокортикоидов при возникновении гемолитических кризов. При всех формах талассемии показан прием препаратов фолиевой кислоты и витаминов группы В.
При гиперспленизме (особенно на фоне гемоглобиноза Н) требуется удаление селезенки (спленэктомия). Из-за склонности к присоединению инфекционных осложнений больным рекомендуется обязательная вакцинация против пневмококковой инфекции. Многообещающим методом лечения талассемии служит трансплантация костного мозга от гистосовместимого донора.
Прогноз больших форм талассемии неблагоприятный; больные погибают в младенческом или молодом возрасте. При гетерозиготной бессимптомной форме талассемии продолжительность и качество жизни в большинстве случаев не страдают. Первичная профилактика талассемии включает предупреждение браков между гетерозиготными носителями генов заболевания, а при высоком генетическом риске рождения больного потомства – отказ от деторождения.
Источник
Талассемии у детей: клиника, диагностика, лечение
Бета-Талассемии наиболее часто встречаются у выходцев с Индийского субконтинента, Средиземноморья и Ближнего Востока. В Великобритании дети, страдающие этими заболеваниями, рождаются от родителей индийского происхождения; в прошлом, многие были рождены от греков-киприотов, однако в настоящее время это происходит редко, поскольку в их сообществе проводится активное консультирование.
Поскольку дефицит синтеза b- и у-цепей продолжается после неонатального периода, производство увеличенной доли HbF и продукция 8-цепей увеличивают количество НbА2. Вследствие этого происходит преципитация цепей глобина внутри мембраны эритроцитов, приводящая к гибели клеток в костном мозге (неэффективный эритропоэз) и преждевременному удалению циркулирующих эритроцитов селезёнкой.
Тяжесть течения бета-талассемии зависит от количества присутствующих НbА и HbF.
• Большая бета-талассемия — НbА (а2р2) не может продуцироваться в связи с патологическим геном р-глобина. Наиболее тяжёлая форма.
• Умеренная бета-талассемия — мутации b-глобина позволяют образовываться небольшому количеству HbA и/или большому количеству HbR Более лёгкая, вариабельная.
• Малая бета-талассемия — один нормальный и один патологический гены b-глобина. Асимптомный носитель.
Большая бета-талассемия — аутосомно-рецессивное наследование мутаций в каждом из двух генов бета-глобина (по одному от каждого из родителей).
Клинические особенности талассемии у детей:
• Тяжёлая анемия и желтуха начиная с 3-6 мес жизни.
• Недостаточная прибавка в весе / задержка развития.
• Экстрамедуллярный гемопоэз, приводящий к распространению костного мозга, что обусловливает признак классического лица с гипертрофией верхней челюсти и выступающим лобным гребнем; отмечается значительная гепатоспленомегалия (не наблюдается у детей, которым проводилась адекватная трансфузия).
Лечение талассемии у детей
Это состояние в целом приводит к смерти без регулярных гемотрансфузий, поэтому всем пациентам проводятся пожизненные ежемесячные трансфузии эритроцитарной массы.
Целью является поддержание концентрации Hb выше 10 г/дл для того, чтобы уменьшить отставание в развитии и предотвратить деформацию костей. Повторные гемотрансфузий приводят к хронической перегрузке железом, которая вызывает сердечную недостаточность, цирроз печени, диабет, бесплодие и отставание в развитии. Для того чтобы минимизировать этот риск, терапия хелатами железа с помощью подкожных инъекций дезферриоксамина, проводимая в ночь перед трансфузией, начинается с 2-3-летнего возраста.
Пациенты, которые хорошо отвечают на трасфузию и хелацию, имеют 90% шанс на то, что они доживут до сорока лет и старше. Однако сложно достигнуть комплайнса. Те, которые не могут переносить лечение, имеют высокую смертность в раннем детстве вследствие перегрузки железом. Альтернативным лечением большой бета-талассемии является трансплантация костного мозга, которая в настоящее время является единственным способом излечения.
Она обычно оставляется в резерве для детей, имеющих братьев или сестёр, идентичных по HLA (главный комплекс гистосовместимости), поскольку в таком случае имеется 90% шансов на успех (т.е. независимость от трансфузий и долгосрочное излечение), и в то же время 5% шансов смерти в связи с трансплантацией.
Осложнения долговременной трансфузии у детей при бета-талассемии:
• Отложение железа — наиболее важное (у всех пациентов):
— Сердце — кардиомиопатия.
— Печень — цирроз.
— Поджелудочная железа — диабет.
— Гипофиз — задержка развития и сексуального созревания.
— Кожа — гиперпигментация.
• Образование антител (10% детей):
— Алло-антитела у пациента сильно осложняют подбор совместимой группы крови.
• Инфекции — в настоящее время нетипичны (<10% детей):
— Прионные болезни (например, новый вариант болезни Крейцфельда-Якоба).
— ВИЧ.
— Гепатит А, В, С.
— Малярия.
• Венозный доступ (распространённая проблема).
— Могут потребоваться приспособления для центрального венозного доступа (например, Portacath — катетер для портальной вены), они создают предрасположенность для инфекций.
— Часто травматичный у маленьких детей.
Пренатальная диагностика бета-талассемии. У обоих родителей, гетерозиготных по бета-талассемии, существует риск 1:4 того, что ребёнок будет страдать этим заболеванием. Необходимо предложить пренатальную диагностику бета-талассемии (анализ ДНК в биоптате ворсин хориона) наряду с генетическим консультированием, для того чтобы оказать помощь родителям принять разумное решение — оставлять беременность или прервать её.
Малая бета-талассемия. У гетерозиготных индивидуумов, как правило, нет симптомов заболевания. Обычно отмечается гипохромия и микроцитоз эритроцитов. Анемия лёгкая или отсутствует с непропорциональным снижением МСН (до 18-22 фл) и MCV (до 60-70 фл). Поэтому количество эритроцитов обычно увеличено (>5,5х1012/л). Наиболее важной диагностической особенностью является увеличение НbА2 (и приблизительно в половине случаев отмечается умеренное увеличение уровня HbF на 1-3%) на электрофорезе Нb.
Лёгкая бета-талассемия может быть спутана с железодефицитной анемией лёгкой степени, что приводит к ненужной терапии железом.
Альфа-талассемии у детей
У здоровых индивидуумов имеются четыре гена альфа-глобина. Проявления синдромов а-талассемии зависят от количества функционирующих генов этого белка.
Наиболее тяжёлая а-талассемия, большая альфа-талассемия (также известная как Нb Барте), возникает в результате делеции всех четырёх генов альфа-глобина, поэтому не может производиться никакого HbA (a2p2). Она обычно встречается в семьях родом из Юго-Западной Азии и проявляется в среднем триместре в форме водянки плода (отёк и асцит) вследствие анемии плода, которая всегда заканчивается гибелью во внутриутробном периоде или в течение нескольких часов после рождения.
Долгосрочное выживание при большой а-талассемии отмечается лишь у тех, кому проводились ежемесячные внутриутробные трансфузии до рождения с последующими пожизненными ежемесячными трансфузиями после рождения. Диагноз устанавливается с помощью электрофореза Нb или высокоэффективной жидкостной хроматографии (ВЭЖХ), на которой выявляют только Нb Барте.
Если происходит делеция только трёх генов а-глобина (болезнь НbН), у детей с таким состоянием отмечается лёгкая/умеренная анемия, но некоторые пациенты нуждаются в трансфузии.
Делеция одного или двух генов а-глобина (известная как лёгкая альфа-талассемия) обычно бессимптомная и анемия лёгкая или отсутствует. Могут быть гипохромия и микроцитарность эритроцитов, поэтому её можно спутать с железодефицитным состоянием.
— Также рекомендуем «Анемия у новорожденных: причины, диагностика»
Оглавление темы «Заболевания крови детей»:
- Талассемии у детей: клиника, диагностика, лечение
- Анемия у новорожденных: причины, диагностика
- Недостаточность костного мозга у ребенка: причины. Анемия Фанкони и синдром Швахмана-Даймонда
- Патологические кровотечения у детей: причины, диагностика
- Гемофилия у детей: диагностика, лечение
- Болезнь Виллебранда у детей: диагностика, лечение
- Тромбоцитопении у детей: причины, диагностика, лечение
- Диссеминированное внутрисосудистое свёртывание (ДВС) у детей: причины, диагностика, лечение
- Тромбозы у детей: причины, диагностика
- Критерии компетентных родителей. Особенности
Источник
Костный мозг у детей с большой талассемией представляет резкую гиперплазию красной кроветворной ткани. Количество эритронормобластов достигает 74,6 — 85,8%, что при наличии выраженной анемии свидетельствует о неэффективном эритропоэзе. Определение щелочеустойчивости гемоглобина и электрофорез его на бумаге обнаруживают у детей увеличение HbF до 50,7 — 88%.
Мы наблюдали следующий случай.
Надя Д., 15 лет, русская.
Диагноз: большая талассемия. Находилась в клинике с 13.ХII.1965 г. по 15.II.1966 т.. Мать страдает аналогичным заболеванием. Девочка родилась весом 3 кг. Больна с рождения. Росла ослабленным ребенком. При поступлении в клинику отмечались бледность, субиктеричность кожи. Изменения в скелете, увеличение селезенки, НЬ 6,6 г%, эрит. 2 660 000, цвет. пок. 0,7, ретик. 58 %0. Осмотич. резист. эрит. 0,4 — 0,24%, резкие анизоцитоз, анизохромия, пойкилоцитоз, мишеневидные эритроциты. Фетальный гемоглобин увеличен до 50,7%. Эритробластический росток костного мозга 85,5%.
Биохимические показатели крови аналогичны таковым при большой талассемии. Количество HbF у больных 10,6 — 24%.
Малая талассемия обычно отличается относительно легким течением гемолитического процесса. Дети чаще всего бывают удовлетворительного физического развития с нормальным весом и ростом. Клинические симптомы заболевания слабо выражены, а иногда и «стерты», поэтому постановка правильного диагноза затруднена. Мы наблюдали ребенка, который в течение 10 лет считался здоровым и только на 11-м году у него была диагностирована малая талассемия с дебюта острого гемолитического криза. Больные с малой талассемией бывают нерезко бледными. Желтуха у них часто отсутствует, селезенка не прощупывается или определяется у края реберной дуги. Только в редких случаях малая талассемия может протекать тяжело во время гемолитического криза.
Средняя талассемия характеризуется более легким клиническим течением в сравнении с большой талассемией. В физическом развитии дети с этой формой заболевания отстают менее резко, а желтуха, бледность и гепатолиенальный синдром не столь выражены. Изменения в скелете у больных приближаются к таковым при большой талассемии. Гематологические показатели характеризуются нерезкой анемией с НЬ 48 — 60 ед., эритроциты 2 — 3 млн. в 1 мм3. Количество лейкоцитов колеблется в пределах 3600 — 11750. В лейкограмме наблюдается увеличение базофилов до 2 — 3%. Количество ретикулоцитов находится на уровне 27 — 95%. Средний диаметр эритроцитов 7,9 — 8 мкм, осмотическая резистентность повышена (0,44 — 0,28%), наблюдается нерезкая тромбоцитозе.
У детей с малой талассемией гематологические показатели лучше, чем у детей, страдающих большой и средней талассемиями. Средний диаметр эритроцитов 7 — 7,5 мкм, осмотическая резистентность эритроцитов повышена. В мазках периферической крови единичные мишеневидные эритроциты. Содержание HbF 2,39 — 45%. У 5 детей наблюдалось увеличение содержания НЬА2, что считается патогномоничным для малой талассемии. Как и в предыдущих группах, у детей отмечалась гипохолестеринемия. В протеинограмме наблюдалось некоторое увеличение фракций глобулинов.
Минимальная талассемия протекает бессимптомно. Она выявляется при специальном обследовании, чаще всего у родителей, больных гетерозиготными формами талассемии. Эти люди обычно являются носителями патологического гена при отсутствии у них клинических симптомов болезни.
«Гемолитические анемии у детей»,
М.Я.Студеникин, А.И.Евдокимова
Сущность лекарственного гемолиза эритроцитов при дефиците Г6ФД заключается в следующем. При встрече эритроцитов с гемолизирующим веществом окислительного действия в норме происходит компенсаторное усиление активности Г6ФД для поддержания редукции глютатиона на необходимом уровне. При наследственном дефиците Г6ФД резервные возможности эритроцитов снижаются и поэтому при встрече их с гемолизирующими агентами резко нарушается процесс восстановления глютатиона, что приводит…
В настоящее время привлекают внимание заболевания, связанные с наследственной недостаточностью ферментов — энзимопатии. Открыто много внутри-эритроцитарных ферментов, недостаток которых может способствовать появлению гемолитической анемии. Ферментопатии можно разделить на две группы. К первой группе относятся дефициты пируваткиназы, глюкозофосфатизомеразы, триозофосфатизомеразы, гексокиназы, фосфофруктокиназы, АТФазы и др. При дефиците ферментов этой группы у больных нарушается течение внутриклеточных гликолитических процессов…
Артур Г., 10 лет. Поступил в клинику 4.1 1971 г. Отец армянин, мать — азербайджанка. У родителей умеренная гипохромная анемия. Мальчик родился весом 3550 г. С раннего возраста страдает экссудативным диатезом. В возрасте 4 лет впервые выявлена анемия. При поступлении в клинику состояние средней тяжести, бледный, склеры субиктеричные, изменения в скелете — «башенный» череп, прогнатизм,…
При гемоглобинозах Н и Zurich чаще всего после дачи лекарственных препаратов развивается гемолитический криз и иногда очень тяжелый. При этом моча может приобретать темный цвет за счет выделения дипирроллов мезабилифусциновой группы. По органам удается выявить спленомегалию. Диагностика гемоглобинопатий группы нестабильных гемоглобинов проводится с помощью лабораторных методов исследования. Они включают достаточно большой арсенал методик, способствующих не…
У большинства гемоглобинов этой группы в каком-то положении р-цепей имеется замена одних аминокислотных остатков на другие. Это приводит к изменению электрофоретической подвижности (преимущественно они мигрируют между гемоглобинами A и А2), нарушению соединения гема с глобином (НЬН Hammersmith, Koln), увеличению образования метгемоглобина (in vivo и in vitro), легкой денатурации белка под влиянием некоторых химических веществ (сульфопрепаратов,…
Источник

