Тромботические микроангиопатические гемолитические анемии
Тромботическая микроангиопатия – клинический синдром, для которого характерны:
- тромбоцитопения;
- микроангиопатическая гемолитическая анемия (неиммунная гемолитическая анемия с шистоцитами (фрагментированные эритроциты) в мазке крови);
- микроваскулярный тромбоз концевых артериол и капилляров с множественной дисфункцией органов.
Морфологически тромботическая микроангиопатия (ТМА) определяется как уплотнение сосудистой стенки с набуханием или отделением эндотелиальных клеток от базальной мембраны и отложением гиалиновых депозитов в субэндотелиальном пространстве, внутрисосудистые тромбоцитарные тромбы и окклюзия сосудов [1]. Повреждение эндотелия сосудов при ТМА индуцирует процесс образования внутрисосудистых тромбоцитарных тромбов мелких сосудов. Потребление тромбоцитов приводит к развитию тромбоцитопении, сужение просвета сосудов вызывает микроангиопатическую гемолитическую анемию (происходит механическое разрушение эритроцитов), ишемию важнейших органов.
К ТМА относят:
- тромботическую тромбоцитопеническую пурпуру,
- гемолитико-уремический синдром,
- HELLP-синдром (hemolysis [H – гемолиз], elevated liver enzymes [EL – увеличение печеночных ферментов], low platelet count [LP – низкое количество тромбоцитов]), возникающий при беременности.
Выделяют также ТМА-ассоциированные синдромы:
- ДВС-синдром,
- катастрофический антифосфолипидный синдром,
- злокачественную гипертензию,
- преэклампсию/эклампсию.
ТМА может развиваться:
- при ряде заболеваний/cиндромов (например, при диффузных болезнях соединительной ткани);
- после приема лекарств (тиенопиридины, хинин, хинидин, цитостатики, ингибиторы кальциневрина, оральные контрацептивы и др.);
- после трансплантации органов и тканей.
Обязательными элементами заболеваний, относящихся к ТМА и ТМА-ассоциированным синдромам, являются микроангиопатическая гемолитическая анемия и тромбоцитопения.
МикроАнгиопатическая Гемолитическая Анемия (МАГА) характеризуется:
- снижением гемоглобина;
- шистоцитами в мазке крови;
- ретикулоцитозом;
- гипербилирубинемией (за счет непрямой фракции);
- снижением уровня гаптоглобина;
- отрицательным прямым антиглобулиновым тестом (или отрицательной пробой Кумбса);
- повышением уровня лактатдегидрогеназы (ЛДГ).
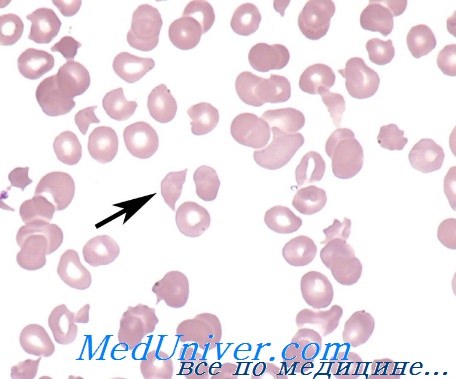
Шистоцитоз (превышение нормального уровня количества шистоцитов) – необходимый диагностический признак тромботической микроангиопатии.
Шистоциты – это фрагменты эритроцитов, выявляемые в мазке крови, в виде:
- полумесяца, с двумя-тремя острыми выступами,
- шлема (каски),
- треугольника,
- маленького неправильной формы фрагмента, имеющего линию разлома [1].
Международный Совет по Стандартизации в Гематологии (ICSH) разработал рекомендации по идентификации шистоцитов. Было предложено также считать шистоцитами микросфероциты (при наличии в мазке крови шистоцитов другой формы) [2].
Диагностические критерии тромботической тромбоцитопенической пурпуры (диагностическая диада) при отсутствии другой выявленной причины:
- Микроангиопатическая гемолитическая анемия (МАГА),
- тромбоцитопения.
Диагностические критерии гемолитико-уремического синдрома (диагностическая триада):
- МАГА,
- тромбоцитопения,
- поражение почек.
Диагностические критерии HELLP-синдрома:
- МАГА,
- тромбоцитопения <100×109/л,
- АСТ (аспартат аминотрансфераза) >70 ед./л.
Л.Б. Филатов,
- Lesesve J.-F., Salignac S., Bordigoni P., Lecompte T., Troussard X. et le Groupe français d’hématologie cellulaire Rôle du biologiste confronté à une recherche de schizocytes. Hématologie 2007; 13(3): 193-204.
Bain B.J. Interactive haematology imagebank. CD. – Blackwell Science, 1999.
Bull B.S., Kuhn I.N. The production of schistocytes by fibrin strands (a scanning electron microscope study). Blood 1970; 35(1): 104-11. - Zini G., d’Onofrio G., Briggs C., et al. ICSH recommendations for identification, diagnostic value, and quantitation of schistocytes. Int. J. Lab. Hematol. 2012; 34(2): 107-16.
Статьи по тромботическим микроангиопатиям:
Макрососудистый тромбоз у тяжелых больных с тромботическими микроангиопатиями
Camous L, Veyradier A, Darmon M, Galicier L, Mariotte E, Canet E, Parquet N, Azoulay E
Тромботическая микроангиопатия, связанная с уровнем сиролимуса после аллогенной трансплантации гемопоэтических клеток с профилактикой РТПХ такролимусом / сиролимусом
Shayani S, Palmer J, Stiller T, Liu X, Thomas SH, Khuu T, Parker PM, Khaled SK, Forman SJ, Nakamura R
Микроангиопатическая гемолитическая анемия, ассоциированная с опухолью: клинические и лабораторные особенности 168 случаев
Lechner K, Obermeier HL
Системные злокачественные новообразования как причина внезапной микроангиопатической гемолитической анемии и тромбоцитопении
George JN
Источник
Микроангиопатическая гемолитическая анемия — история изучения, причиныМикроангиопатическая гемолитическая анемия (МГА) относится к группе приобретенных гемолитических анемий по внеэритоцитным причинам, точнее — к подгруппе механических гемолитических анемий. Микроангиопатическая гемолитическая анемия представляет собой частную форму гемолитической анемии, характеризующуся наличием раздробленных эритроцитов («шлемообразные », треугольные, зубчатые, микросфероциты) на мазках крови и признаками внутрисосудистого расплавления крови. Часто тромбоцитопения и расстройство свертывания (Brain) сопровождают микроангиопатическую гемолитическую анемию. Подобно иным видам анемии, микроангиопатическая гемолитическая анемия не составляет самостоятельную единицу, а лишь синдром, развивающийся при ином заболевании или синдроме. Болезни, при которых была описана микроангиопатическая гемолитическая анемия носят общий характер — патологическое изменение небольших сосудов, артериол и капилляров (микроаигиопатия). В 1891 г. Ehrlich впервые отметил наличие раздробленных эритроцитов («шистоциты») на мазке страдающего анемией. В 1949 Schwartz и Motto описали присутствие 0,1—0,5% «надрезанных» («burr cells») эритроцитов в мазках крови больных уремией, раком желудка и пептической геморрагической язвой. В 1954 г. Monroe и Strauss сообщили о выявлении раздробленных эритроцитов на срезах отдельных кровеносных сосудов больного, погибшего от тромботической тромбогемолитической пурпуры. Они выдвинули гипотезу, по которой раздробление эритроцитов якобы происходит в ненормальных кровеносных сосудах. В 1962 Brain, Dacie и Hourihane впервые использовали термин «микроангионатическая гемолитическая анемия». В период с 1962 по 1972 гг. Brain и Dacie, в сотрудничестве с другими исследователями, поставили эксперимент микроангиопатической гемолитической анемии на животных, изучили механизм дробления эритроцитов в пробирке, описали процесс внутрисосудистого свертывания у страдающих микроангиопатической гемолитической анемией, реакцию на лечение гепарином и сочетание микроангиопатической гемолитической анемии с метастатическим раком. Понятие микроангиопатической гемолитической анемии общепринято и подтверждено другими авторами. С 1961 г. в литературе был опубликован ряд сообщений о гемолитической анемии, в условиях которой, после хирургического вмешательства на сердце по поводу клапанопластики, протезирования синтетическими клапанами и исправления внутрисердечных дефектов. Гемолитическая анемия с дроблением эритроцитов описана у больных, страдающих тяжелой недостаточностью клапанов (в частности сужение аорты) и коарктацией аорты.
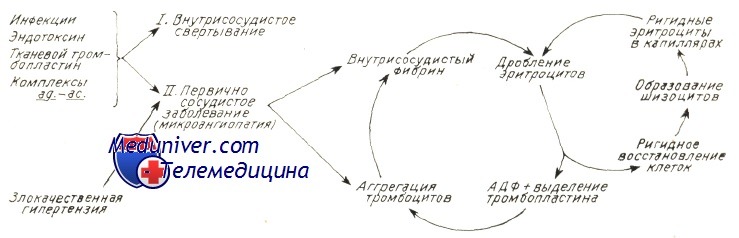
Причины (этиология) микроангиопатической гемолитической анемииМикроангиопатическая гемолитическая анемия описана в сочетании с рядом иных заболеваний. Наличие процесса расплавления крови с последующим дроблением эритроцитов предполагает следующие патогенетические механизмы: В патологии человека микроангиопатическая гемолитическая анемия описана в сочетании с сосудистыми заболеваниями, обусловленными обоими механизмами. В случае первичного сосудистого нарушения или вызванного процессом внутрисосудистого свертывания тромбы фибрина и тромбоциты частично закупоривают просвет мелких сосудов. Волоски фибрина действуют на движущиеся эритроциты подобно «гильотине » и тем самым обусловливают их дробление. Те фрагменты эритроцитов, у которых поверхностная оболочка меньше соответствующего объема, быстро захватываются макрофагами селезенки. Другие же, у которых отношение площадь/ объем превышает норму (за счет перехода гемоглобина в плазму или в результате дробления) подвергаются процессу рубцевания («повторному запечатыванию») оболочки и остаются в кровообращении, представляя собой «ключ» к постановке диагноза основного заболевания. В результате этого процесса рубцевания оболочки появляются жесткие эритроциты — сфероциты, «шлемообразные» эритроциты, которые подвергаются повторному дроблению при переходе через капилляры. Дробление эритроцитов сопровождается выделением АДФ и фосфолипидного прокоагулянта, что снова ведет к аггрегации тромбоцитов и отложению фибрина в мелких сосудах, тем самым усиливая процесс внутрисосудистого свертывания. На рисунке приводится модель патогенеза микроангиопатической гемолитической анемии. Необходимо отметить, что не у всех больных с рассеянным внутрисосудистым свертыванием наблюдается микроангиопатическая гемолитическая анемия. Для ее развития важно, чтобы фибрин удержался в кровообращении достаточный период времени. Сохранение запасов фибрина в мелких сосудах зависит от ритма дефибринизации, клиренса макрофаговой системы и местного фибринолиза.
Болезни, сопутствующие микроангиопатической гемолитической анемии (МГА)1. Тромбогемолитическая тромботическая пурпура (РТТ) 2. Уремический гемолитический синдром При микроангиопатической гемолитической анемии, встречающейся в заболеваниях метастазом рака, эритроциты разрушаются механическим путем в малых сосудах, измененных: — Также рекомендуем «Диагностика микроангиопатической гемолитической анемии — дифференциация» Оглавление темы «Гемолитические анемии»:
|
Источник
Введение
Преэклампсия и HELLP-синдром являются грозным осложнением беременности. В настоящее время они рассматриваются как варианты тромботической микроангиопатии (ТМА). Наиболее грозным представителем ТМА является атипичный гемолитико-уремический синдром (аГУС), к развитию которого предрасполагают генетические аномалии в системе иммунитета. Установлено, что беременность сама по себе может активировать патологический иммунный ответ, причем выраженность активации возрастает при наличии акушерских осложнений, достигая максимума у пациенток с преэклампсией. Генетический дефект в сочетании с преэклампсией приводит к неконтролируемой активации иммунного ответа, являющегося при акушерском аГУС основой развития полиорганной недостаточности, которая не может быть устранена без специфического лечения.
Критерии диагноза ТМА
ТМА представляет собой синдром, в основе которого лежит повреждение эндотелия сосудов микроциркуляторного русла (МЦР) различными способами, но имеющий сходные проявления и диагностические признаки. Результатом эндотелиального повреждения служит тромботическая микроангиопатия — особый тип поражения мелких сосудов, представленный их тромбозом и воспалением сосудистой стенки.
Морфологическая картина ТМА: отек эндотелиальных клеток, их отслойка от базальной мембраны (эндотелиоз), некроз, деструкция, расширение субэндотелиального пространства, тромбы в просвете капилляров и артериол, содержащие тромбоциты и фибрин, нередко с полной окклюзией просвета сосудов.
Клинико-лабораторные признаки ТМА:
- Микроангиопатическая гемолитическая анемия (МАГА): (Кумбс—негативная гемолитическая анемия с высоким уровнем ЛДГ, низким уровнем гаптоглобина и наличием шизоцитов в мазке периферической крови).
- Тромбоцитопения (потребления).
- Ишемическое поражение органов (почек, ЦНС и др.).
Тромботические микроангиопатии классифицируют на первичные и вторичные.
Первичные ТМА:
- Тромботическая тромбоцитопеническая пурпура (ТТП) — в основе которой лежит дефицит фермента ADAMTS-13 (активность менее 10%).
- Типичный ГУС (инфекционно-опосредованный), вызываемый бактериями, продуцирующими шигатоксин (STx), в первую очередь, E.coli (STEC-ГУС).
- Атипичный ГУС — обусловлен генетическими нарушениями регуляторных белков системы комплемента.
Вторичные ТМА вследствие следующих состояний:
- Беременность и роды: преэклампсия/эклампсия, HELLP-синдром.
- Аутоиммунные заболевания: системная красная волчанка (СКВ), системная склеродермия, антифосфолипидный синдром (АФС).
- Злокачественные опухоли.
- Инфекции, в том числе ВИЧ, грипп А (H1N1), сепсис, септический шок.
- Злокачественная артериальная гипертензия, гломерулопатии.
- Метилмиалоновая ацидурия с гомоцистеинурией.
- Лекарственная терапия: хинин, интерферон, ингибиторы кальциневрина (циклоспорин, такролимус), ингибиторы mTOR (сиролимус, эверолимус), противоопухолевые препараты (цисплатин, гемцитабин, митомицин, ингибиторы VEGF и тирозинкиназы — бевацизумаб, сунитинаб, сорафениб), пероральные контрацептивы, валациклавир.
- Ионизирующее излучение.
- Трансплантация органов и костного мозга.
Критерии диагноза аГУС в акушерстве
Диагноз аГУС в акушерстве — это диагноз исключения. Дифференциальная диагностика с другими формами ТМА приведена в табл. 1.
Таблица 1
Заболевание | Дифференциально-диагностические признаки |
Типичный ГУС | Положительный результат при бактериологическом исследовании кала: посев на среду для выявления STEC (Mac Conkey для 0157:Н7), определение в образцах фекалий ДНК энтеро-геморрагических E.coli методом ПЦР; выявление в сыворотке антител к липополисахаридам наиболее распространенных в данном регионе серотипов E.coli. |
Наследственная или приобретенная ТТП | Дефицит ADAMTS-13 — активность менее 10%, антитела к ADAMTS-13 |
Беременность. Исключить преэклампсию и HELLP-синдром | Ферменты печени, срок гестации, критерии преэклампсии и тяжелой преэклампсии, положительная динамика непосредственно после родоразрешения |
Аутоиммунные заболевания (системная красная волчанка, антифосфолипидный синдром) | Анти-ДНК-антитела, антинуклеарные антитела, антитела к кардиолипину IgG и/или IgM изотипов, антитела к (32 GP 1 IgG и/или IgM изотипов с помощью стандартизованного иммуноферментного метода, волчаночный антикоагулянт стандартизованным коагулологическим методом |
ВИЧ-инфекция | Положительные результаты иммунного блоттинга на ВИЧ- инфекцию |
Сепсис | Наличие очага инфекции и полиорганной недостаточности (острое изменение по шкале SOFA >2 баллов)| |
Генетическое исследование и биопсия почки не являются необходимыми для установления диагноза аГУС и не играют роли для решения вопроса о тактике лечения больного.
Преэклампсия и HELLP-синдром являются специфическими ассоциированными с беременностью формами ТМА. Всем пациенткам, госпитализированным с диагнозом тяжелая преэклампсия и/или HELLP-синдром, необходимо до родоразрешения исследовать ЛДГ, гаптоглобин в сыворотке крови и шизоциты в мазке периферической крови, а также определить количество тромбоцитов и уровень креатинина.
Истинные тяжелая преэклампсия и HELLP-синдром требуют родоразрешения с целью элиминации секретирующегося анти-ангиогенного фактора sFlt-1 плаценты.
Поскольку термин «HELLP-синдром» — собирательное понятие и его причины до конца не выяснены, тактика родоразрешения и интенсивной терапии строится в соответствии с тактикой при тяжелой преэклампсии (родоразрешение). В этом случае диагноз формулируется в соответствии с МКБ Х — «Тромботическая микроангиопатия (HELLP-синдром)».
Принципы и схемы терапии
При развитии клиники HELLP-синдрома в послеродовом периоде необходимо строить тактику интенсивной терапии в зависимости от следующих клинических вариантов:
Вариант 1. У пациентки сохранены: сознание, диурез более 0,5 мл/кг/ч (вне зависимости от цвета мочи), стабильная гемодинамика (или с тенденцией к артериальной гипертензии), отсутствует геморрагический синдром любой локализации. При лабораторном исследовании выявлены тромбоцитопения, повышены уровни АСТ, АЛТ, ЛДГ, массивного внутрисосудистого гемолиза нет. Плазменные факторы свертывания в норме. В данном случае, в течение 1–3 суток оценивается динамика клинико-лабораторных
проявлений HELLP-синдрома и при отсутствии отрицательных проявлений интенсивная терапия ограничивается базовой терапией преэклампсии и инфузией кристаллоидов 15–20 мл/кг/сутки. Пациентка получает нутритивную поддержку и активизируется. Проводится тромбопрофилактика НМГ при количестве тромбоцитов более 70000 в мкл.
Вариант 2. Уже с первых часов после родоразрешения развивается клиника острой печеночной недостаточности (тромбоцитопения, рост АСТ, АЛТ, коагулопатия, кровотечение, шок, ОПН, ОРДС и т.д.), в основе, которой лежит некроз печени (подкапсульная гематома). Требует проведения комплексной посиндромной интенсивной терапии острой печеночной недостаточности в условиях многопрофильного ЛПУ с возможностью хирургического лечения.
Вариант 3. Развитие массивного внутрисосудистого гемолиза (свободный гемоглобин в крови и моче, анемия) уже в первые часы осложняется развитием ОПН (по шкалам RIFLE, AKIN, KDIGO) и требует проведения заместительной почечной терапии. Противопоказано применение магния сульфата и инфузионной терапии. Требует проведения комплексной посиндромной интенсивной терапии ОПН в условиях многопрофильного ЛПУ. При сохранении или прогрессировании симптомов ТМА (тромбоцитопения и МАГА) в течение 48 часов следует, как один из вероятных диагнозов рассматривать аГУС и проводить соответствующую терапию.
Вариант 4. В исключительных случаях верификации диагноза ТТП в послеродовом периоде на основании сочетания признаков HELLP-синдрома, нарастающей тромбоцитопении, симптомов поражения почек и/или ЦНС со снижением активности ADAMTS-13 менее 10% показана инфузия свежезамороженной плазмы и проведение плазмообмена«.
Вариант 5. Женщинам, перенесшим акушерскую ТМА (преэклампсия, HELLP-синдром), следует устанавливать диагноз аГУС, если после родоразрешения их состояние не улучшается или ухудшается, в короткие сроки (48–72 часов) приводя к формированию
полиорганной недостаточности, что свидетельствует о персистировании ТМА с генерализацией микроангиопатического процесса.
В первую очередь о возможном аГУС следует думать при развитии тяжелого HELLP-синдрома с признаками внепеченочного поражения, особенно если родоразрешение не сопровождается положительной динамикой состояния пациентки, несмотря на лечение в соответствии с клиническими рекомендациями (протоколом лечения) МЗ РФ «Гипертензивные расстройства во время беременности, в родах и послеродовом периоде. Преэклампсия. Эклампсия» 2016 г. Быстрое нарастание анемии при отсутствии выраженной кровопотери свидетельствует об усилении микроангиопатического гемолиза, что, как правило, сопровождается усугублением тромбоцитопении и стремительным ухудшением функции почек, приводящим к развитию олигурической.
Родильницам с установленным диагнозом аГУС следует назначать патогенетическую терапию, направленную на блокирование С5-компонента системы комплемента, играющего ключевую роль в развитии данного осложнения. Антикомплементарная терапия проводится согласно рекомендациям по лечению аГУС взрослых.
Экулизумаб — рекомбинантное гуманизированное моноклональное антитело класса Ig G к С5 компоненту комплемента. Препарат блокирует расщепление С5, препятствуя образованию мембрано-атакующего комплекса и предотвращая тем самым повреждение эндотелия и, следовательно, прекращая процессы микроциркуляторного тромбообразования. Применение Экулизумаба приводит к обратному развитию ТМА и/или предупреждает прогрессирование поражения почек. Критериями эффективности терапии Экулизумабом являются прекращение микроангиопатического гемолиза (снижение уровня ЛДГ до нормальных значений) и нормализация числа тромбоцитов, а также улучшение функции почек.
Начальный курс терапии Экулизумабом рассчитан на 5 недель, далее подразумевается переход на цикл поддерживающего лечения.
Индукционный курс: 1 раз в неделю вводят по 900 мг Экулизумаба на протяжении 4-х недель. На пятой неделе дозу увеличивают до 1200 мг.
Поддерживающий этап: каждые 14 (плюс/минус 2 дня) дней вводят по 1200 мг.
В ожидании Экулизумаба родильницы с установленным диагнозом аГУС должны получать в случае необходимости почечную заместительную терапию при наличии ОПН. Свежезамороженная плазма в больших объемах у пациенток с тяжелой преэклампсией может вызвать перегрузку объемом (TACO-синдром) или развитие иммунного TRALI-синдрома и в отсутствие клинических проявлений коаулопатии и кровотечения противопоказана! Свежезамороженная плазма применяется только при верификации диагноза ТТП.
Ключевые рекомендации
- Акушерский аГУС ассоциирован с высоким риском материнской и перинатальной смертности, неблагоприятным общим и почечным прогнозом.
- Подозрение на акушерскую ТМА требует дифференциальной диагностики между аГУС, ТТП, преэклампсией, HELLP-синдромом, КАФС, острой жировой печенью беременных для выбора тактики лечения. Акушерская ТМА — важная причина синдрома полиорганной недостаточности при беременности и после родов.
- Возможна манифестация акушерского аГУС развернутыми признаками HELLP-синдрома. Напротив, ранний дебют аГУС может привести к развитию преэклампсии.
- Преэклампсия и HELLP-синдром являются специфическими, ассоциированными с беременностью, формами ТМА. Всем пациенткам, госпитализированным с диагнозом преэклампсия и/или HELLP-синдром, необходимо до родоразрешения исследовать лабораторные маркеры ТМА полном объеме (шизоциты, ЛДГ, гаптоглобин, число тромбоцитов), а также определять уровень креатинина сыворотки.
- Если у пациентки с установленным диагнозом HELLP-синдром своевременно начатая адекватная терапия не приводит к его регрессу в течение 48–72 часов, следует трансформировать диагноз в аГУС и начинать терапию экулизумабом.
- Акушерский аГУС — сложный диагноз, и для его постановки и выработки тактики лечения необходим междисциплинарный подход и содружественная работа акушеров-гинекологов, анестезиологов-реаниматологов, нефрологов, гематологов

Качушкин Е.Ю.
Другие статьи
Для чего нужен приём препаратов содержащих бифидо и лактобактерии в гинекологии? Ответ можно дать коротким предложением: для восстановления микрофлоры полового канала после лечения воспалительных заболеваний и дисбактериоза.
Можно сказать, что после 40 недель все только начинается, возникают волнения «а почему еще я не родила»? «Что со мной не так»?!
Последнее время мы все чаще встречаемся с маммопластикой, и ,естествено, все женщины переживают, смогут ли они кормить, придет ли у них молоко, не вредны ли импланты для ребенка (не меняют ли они состав грудного молока)!
Источник

