Наследственные гемолитические анемии патофизиология
(для
внутрикафедрального пользования)
МИНИСТЕРСТВО
ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ
УО
«ГОМЕЛЬСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ
УНИВЕРСИТЕТ»
Кафедра
патологической
физиологии
Утверждено
на заседании кафедры
протокол
№
7 от 31.08.2015
Зав.
кафедрой патофизиологии, к.м.н.
доцент_____________Т.С.
Угольник
Учебно-методическая
разработка для студентов
лечебного
факультета
Гомель
2015
Патофизиология системы крови. Гемолитические анемии. Эритроцитозы
Актуальность
темы: Гемолитические
анемии различны по этиологии, механизмам
развития, клинико-гематологической
картине, поэтому изучение этих видов
анемий особенно важно в практической
деятельности врача.
2.
Цель занятия:
изучить этиологию, патогенез и основные
проявления гемолитических анемий.
3.
Задачи занятия: Знать
классификацию, этиологию и патогенез
гемолитических анемий. Изучить основные
нарушения и компенсаторно-приспособительные
процессы в организме при анемиях и
полицитемиях.
Основные учебные вопросы (план):
Патология
системы эритрона. Анемии и эритроцитозы,
определение понятий, принципы
классификации, общая характеристика.Анемии
при лейкозах и других опухолевых
процессах.Наследственные
гемолитические анемии: эритроцитопатии,
эритроэнзимопатии, гемогобинопатии
причины и механизмы развития.Приобретенные
гемолитические анемии: виды, причины,
механизмы развития, проявления в
органах кроветворения и в периферической
крови.Роль
аутоиммунных процессов в патогенезе
анемий. Аутоиммунные гемолитические
анемии.Эритроцитозы
первичные и вторичные: причины, механизмы
развития, проявления в органах
кроветворения и в периферической
крови.Нарушения
и компенсаторно-приспособительные
процессы в организме при анемиях и
эритроцитозах.Принципы
терапии анемий.
5. Вспомогательные материалы по теме:
Группа
анемий, наследственно обусловленных
или приобретенных, общим признаком
которых является укорочение жизни
эритроцитов. При этом имеет место стойкое
(хроническая ГА) или массированное
(острая ГА) преобладание разрушения
эритроцитов над их образованием.
Проявляется заболевание синдромами
усиленного гемолиза и компенсаторного
усиления эритропоэза. Усиление гемолиза
(гемолитические кризы) наблюдается при
всех ГА и нередко развивается после
интеркуррентных заболеваний, большой
физической нагрузки, в результате
стрессов, интоксикаций и т. д. В ряде
случаев провоцирующий агент установить
не удается.
Классификация га:
I. Наследственные:
– эритроцитопатии
(мембранопатии);
– ферментопатии
(энзимопатии);
– гемоглобинопатии
(гемоглобинозы).
II. Приобретенные:
Иммунные
формы:
изоиммунные
гетероиммунные
формыаутоиммунные
формы
Неиммунные
формы:
токсико-гемолитические
инфекционные
механические
Развитие
наследственных ГА обусловлено внутренними
аномалиями эритроцитов (эндоэритроцитарные);
приобретенных — влиянием факторов,
действующих вне эритроцита
(экзоэритроцитарные).
Гемолиз
эритроцитов
при гемолитических анемиях может
происходить внутриклеточно (так же как
и физиологический гемолиз), или
непосредственно в сосудах. В связи с
этим выделяют 2 типа патологического
гемолиза:
1.
Внутриклеточный гемолиз—
разрушение «маркированных» иммуноглобулином
(Ig) G эритроцитов в РЭС при наследственной
патологии мембраны эритроцитов,
нарушениях активности ферментов, синтеза
гемоглобина, при несовместимости по
эритроцитарным антигенам между матерью
и плодом и при гемотрансфузиях.
2.
Внутрисосудистый гемолиз
— комплементзависимый лизис «маркированных»
IgM (реже IgG) эритроцитов непосредственно
в кровотоке (в сосудах) при действии
каких-либо внешних факторов, которые
вызывают прямое или опосредованное
повреждение клеток. Причиной этого
может быть разрушение мембраны эритроцитов
вследствие механической травмы (при
окклюзии сосудов, гемодиализе, протезах
клапанов сердца и др.), под влиянием
физических (ионизирующая радиация,
высокая температура), токсических (при
действии экзо- и эндотоксинов), инфекционных
и иммунных (при образовании антиэритроцитарных
аутоантител) патологических факторов.
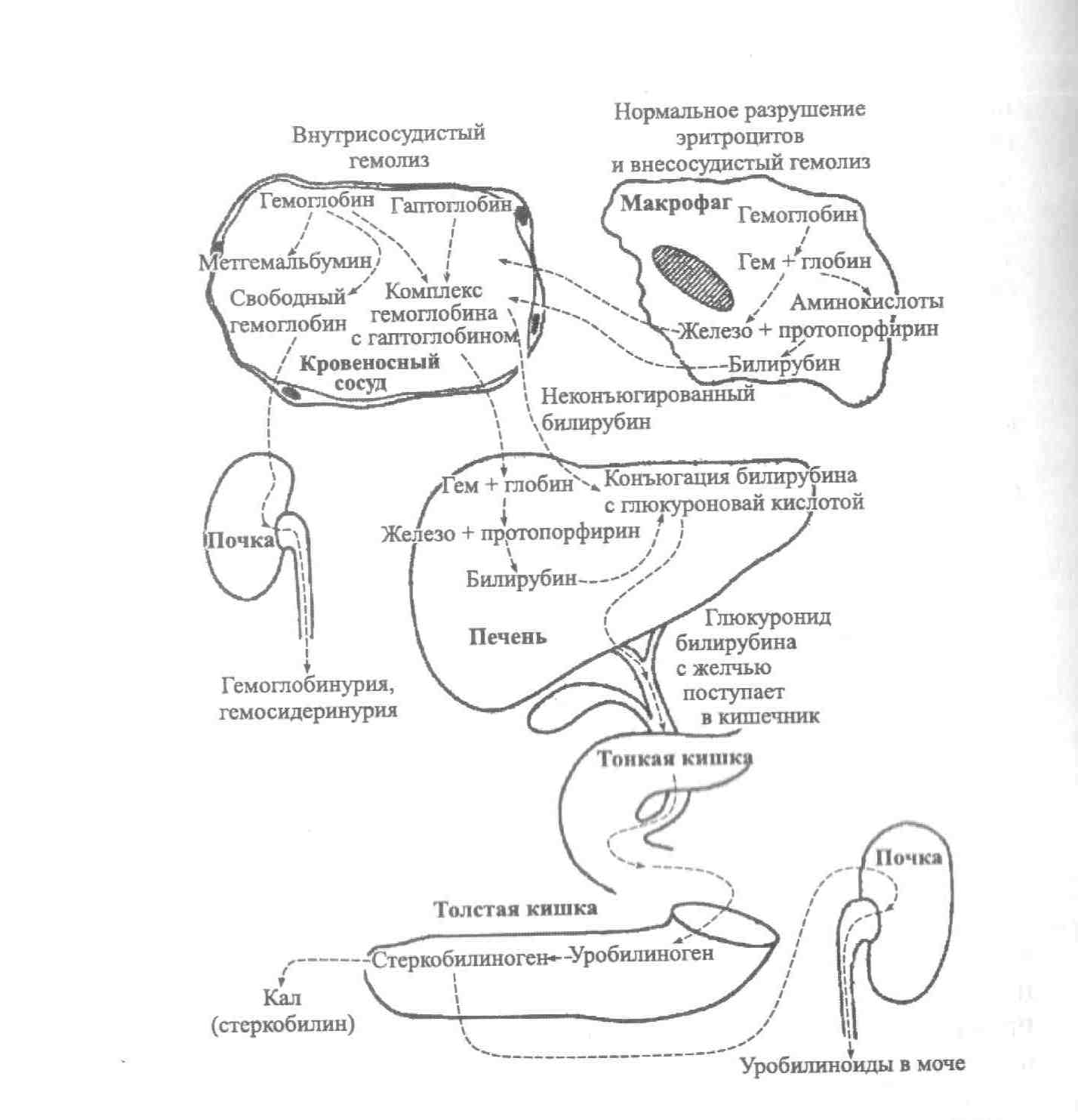
Рисунок
— 1. Механизмы внутрисосудистого и
внутриклеточного гемолиза.
Внутриклеточный
гемолиз
происходит внутри фагоцитов. Из
разрушенных эритроцитов освобождается
гемоглобин, который затем распадается
на глобин, железо и протопорфирин. Глобин
под действием протеолитических ферментов
расщепляется на аминокислоты, с плазмой
они переносятся во внутренние органы
для дальнейшего синтеза белков. Железо
при помощи трансферрина транспортируется
в кровь, затем в костный мозг и в органы,
депонирующие железо (преимущественно
в печень). Протопорфирин превращается
в биливердин, а затем в билирубин
(неконъюгированный билирубин), который
транспортируется альбумином в клетки
печени, где связывается с глюкуроновой
кислотой и метаболизируется в диглюкуронид.
Диглюкуронид выделяется в желчь, а затем
в кишечник, где превращается в уробилиноген,
затем стеркобилиноген и выводится с
мочой (уробилиноиды) и калом.
При
внутрисосудистом
гемолизе
эритроциты распадаются в кровеносном
русле, высвобожденный из эритроцитов
гемоглобин связывается с гаптоглобином
плазмы и транспортируется к клеткам
ретикулоэндотелиальной системы печени,
где происходит дальнейший распад
гемоглобина с образованием свободного
билирубина, который затем подвергается
глюкуронидированию. Не связавшийся с
гаптоглобином гемоглобин выводится с
мочой (гемоглобинурия, гемосидеринурия).
В
результате повышенного гемолиза
эритроцитов в крови накапливается
большое количество непрямого билирубина,
что приводит к развитию желтухи. Помимо
этого главным признаком повышенного
внутриклеточного гемолиза является
увеличение селезенки (спленомегалия),
в случаях внутрисосудистого разрушения
эритроцитов ведущим симптомом становится
появление гемоглобина в моче
(гемоглобинурия), что сопровождается
изменением ее окраски вплоть до черного
цвета (табл. 1).
Таблица
1.
Дифференциальные
признаки внутрисосудистого и
внутриклеточного гемолиза
Признаки | Виды | |
внутрисосудистый | внутриклеточный | |
Локализация | Сосуды | РЭС |
Локализация | Канальцы | Селезёнка, |
Желтушность | Умеренная | Выраженная |
Увеличение | Незначительное | Значительное |
Ведущие | Нормохромная | |
Гемоглобинемия Гемоглобинурия Гемосидеринурия | Гипербилирубинемия Повышенное | |
Все
формы малокровия, связанные с повышенной
гибелью эритроцитов периферической
крови, относятся к группе регенераторных
анемий с нормобластическим типом
эритропоэза.
Гемолитическая
анемия — анемия, возникающая, когда
разрушение эритроцитов преобладает
над их образованием.
Классификация.
По этиологии гемолитические анемии
подразделяются на приобретенные
и наследственные.
В свою очередь в зависимости от
этиологических факторов, вызвавших
гемолиз эритроцитов, приобретенные
гемолитические анемии делятся на
токсические,
обусловленные
действием экзогенных и эндогенных
гемолитических ядов; иммунные (гетеро-,
изо-, аутоиммунные), когда гемолиз
происходит под влиянием комплекса
антиген — антиэритроцитарное антитело;
механические—
при механическом повреждении эритроцитов;
мембранопатии,
связанные с соматической мутацией
пролиферирующих клеток эритроцитарного
ряда и образованием популяции эритроцитов
с дефектом структуры мембраны.
На
основании того, какие генетические
нарушения привели к усилению гемолиза
эритроцитов, наследственные гемолитические
анемии подразделяют на наследственные
мембранопатии, ферментопатии и
гемоглобинопатии,
вызванные генетическими дефектами
структуры мембраны, активности ферментов
эритроцитов и синтеза гемоглобина.
Имеется две разновидности наследственных
гемоглобинопатий: анемии, связанные с
нарушением синтеза цепей глобина, и
анемии, обусловленные наследственным
дефектом первичной структуры цепей
глобина.
Этиология
приобретенных гемолитических анемий.
Токсическая гемолитическая
анемия
может развиться под влиянием гемолитических
ядов (соединения мышьяка, свинца,
нитробензол, фенилгидразин; алкоголь,
желчные кислоты, токсические продукты
азотистого обмена; змеиный, грибной,
пчелиный яды и др.), а также при действии
возбудителей инфекционных и паразитарных
заболеваний (гемолитический стрептококк,
анаэробная инфекция, малярийный
плазмодий, лейшмания).
Иммунная
(гетеро-, изо-, аутоиммунная) гемолитическая
анемия
развивается при переливании несовместимой
крови; резус-несовместимости матери и
плода; образовании аутоантител против
собственных эритроцитов при изменении
их антигенных свойств под влиянием
лекарственных препаратов, вирусов, МО
или в результате соматической мутации
иммуноцитов, когда возникает «запретный»
клон лимфоцитов, продуцирующих антитела
к нормальным антигенам эритроцитов
(при лейкозе, системной красной волчанке
и др.).
Механическое
повреждение эритроцитов
может возникнуть при протезировании
кровеносных сосудов и клапанов сердца,
длительном марше или беге по твердому
грунту (маршевая гемоглобинурия),
спленомегалии.
Причиной
приобретенной
мембранопатии
может стать соматическая мутация
эритробластов под действием вирусов,
МО, лекарственных препаратов с образованием
патологической
популяции эритроцитов, у которых
нарушается структура мембраны и
повышается чувствительность к комплементу
(паро- ксизмальная ночная гемоглобинурия).
Патогенез.
Механизм гемолиза при приобретенной
гемолитической анемии
заключается в повреждении структуры
мембран эритроцитов. Одни гемолитические
факторы (например, механические) оказывают
прямое повреждающее действие, другие
(мышьяковистый водород, нитриты), являясь
сильными окислителями, вызывают сначала
метаболические, а затем функциональные
и структурные изменения в мембране и
строме эритроцитов, приводящие к их
гемолизу. Многие гемолитические яды
биологического происхождения обладают
ферментной активностью (лецитиназная
активность стрепто-, стафилолизинов,
яда насекомых и змей), разрушая лецитин
мембран. При иммунных гемолитических
анемиях IgG
и IgM
присоединяют к эритроцитарной мембране
комплемент, который при этом активируется
и вызывает ее ферментативный лизис.
Под
влиянием гемолитических агентов в
мембранах эритроцитов образуются поры,
через которые из клетки выходят ионы
калия, фосфаты, а ионы натрия поступают
в клетку. Вследствие сдвигов ионного
баланса вода проникает в эритроцит,
который при этом набухает, приобретает
сферическую форму, его клеточная
поверхность уменьшается, снижается
способность к деформации. Такие сфероциты
не могут пройти через межэндотелиальные
поры синусов селезенки и фагоцитируются
селезеночными макрофагоцитами. Когда
объем эритроцита достигает критического
(146 % первоначального), а размер пор
мембраны превышает 6 нм, наступает
гемолиз с выходом гемоглобина в плазму.
Гемолиз
эритроцитов при приобретенных
гемолитических анемиях происходит
преимущественно в кровеносном русле.
Однако при резус- конфликте (гемолитическая
болезнь новорожденных) антирезусные
агглютинины, образовавшиеся в организме
резус-отрицательной матери, вызывают
гемолиз резус-положительных эритроцитов
плода или новорожденного не только
внутри сосудов, но и в печени и селезенке
(внутриклеточный гемолиз).
При
наследственной
гемолитической
анемии гемолиз обусловлен снижением
осмотической и механической резистентности
эритроцитов с генетически детерминированными
нарушениями структуры мембраны,
метаболизма, синтеза гемоглобина.
Так,
при наследственной
мембранопатии (микросфероцитарная
гемолитическая анемия или болезнь
Минковского—Шоффара с аутосомно-доминантным
наследованием) генетический дефицит в
мембране эритроцитов Сап-зависимой
АТФазы и фосфолипидов приводит к
повышению проницаемости мембраны. В
клетки поступают ионы натрия и вода,
эритроциты превращаются в сфероциты
с резко пониженной способностью
деформироваться при прохождении через
синусы селезенки. Отрыв части оболочки
у таких эритроцитов ведет к образованию
микросфероцитов с укороченной
продолжительностью жизни (8 — 14 дней
вместо 120 дней в норме) в связи с захватом
их макрофагоцитами селезенки и печени
(внутриклеточный гемолиз).
При
наследственной
ферментопатии,
например глюкозо-6- фосфатдегидрогеназодефицитной
анемии (доминантное, сцепленное с X-
хромосомой наследование), острый
внутрисосудистый гемолиз эритроцитов,
возникающий при приеме лекарств с
высокой окислительной способностью
(противомалярийные препараты, фтивазид
и др.), обусловлен повреждением клеточных
мембран перекисями, так как в эритроцитах
с дефицитом Г-6-ФДГ понижено содержание
восстановленного глутатиона
(антиоксиданта).
Внутриклеточный
гемолиз эритроцитов при наследственной
гемоглобинопатии
связан с синтезом аномального или не
свойственного данному возрасту
гемоглобина. Так, при серповидно-
клеточной анемии образуется HbS
(в (3-цепи глобина глутаминовая кислота
заменена валином), который в восстановленном
состоянии выпадает в кристаллы и вызывает
деформацию эритроцитов (серповидная
форма); гипоксия способствует усилению
гемолиза таких эритроцитов. Следствием
массивного гемолиза эритроцитов является
анемия с нарушением дыхательной функции
крови и развитием гипоксии.
Образовавшийся
при распаде эритроцитов гемоглобин
циркулирует в крови (гемоглобинемия) и
соединяется с гаптоглобином в
крупномолекулярный комплекс, не
проходящий через почечный фильтр. Если
же содержание свободного гемоглобина
в плазме превышает 20,9 ммоль/л (337 г/л) или
исходный уровень гаптоглобина низкий,
тогда не связанный с последним гемоглобин
начинает выделяться с мочой (гемоглобинурия).
Частично гемоглобин поглощается клетками
макрофагоцитарной системы и расщепляется
в них до гемосидерина. Гемосидероз
селезенки, почек, печени, костного мозга
сопровождается реактивным разрастанием
соединительной ткани и нарушением
функций этих органов. Повышенное
образование из гемоглобина желчных
пигментов обусловливает развитие
гемолитической
желтухи.
Кроме того, внутрисосудистый распад
эритроцитов может привести к появлению
тромбов и нарушению кровоснабжения
тканей, отсюда — трофические язвы
конечностей, дистрофические изменения
в селезенке, печени, почках. В результате
поступления в сосудистое русло большого
количества эритроцитарного тромбопластина
возможно развитие ДВС-синдрома.
Картина
крови.
Приобретенная гемолитическая анемия
по типу кроветворения является
эритробластической, по степени регенерации
костного мозга — регенераторной, по
цветовому показателю — нормо- или
гипохромной, реже — ложногиперхромной
(вследствие абсорбции гемоглобина на
эритроцитах). Степень уменьшения
количества эритроцитов и гемоглобина
зависит от интенсивности гемолиза. В
мазке крови обнаруживаются клетки
физиологической регенерации и
дегенеративно измененные эритроциты
(пойкилоцитоз; разорванные, фрагментированные
эритроциты, анизоцитоз). Появление
большого количества эритробластов и
нормобластов характерно для гемолитической
болезни новорожденных.
При
наследственной гемолитической анемии
отмечается усиленная регенерация
эритроцитарного ростка часто с
неэффективным
эритропоэзом,
когда в костнбм мозге разрушаются
ядерные формы эритроцитов. В мазке крови
наряду с регенеративными формами
(высокий ретикулоцитоз, полихроматофилия,
единичные ядерные формы эритроцитов)
находятся дегенеративно измененные
клетки (микросферо- циты при болезни
Минковского — Шоффара, серповидные при
S-
гемоглобинопатии, мишеневидные,
базофильно пунктированные — при
талассемии). При частых гемолитических
кризах может возникнуть гипорегенераторная
анемия.
Соседние файлы в предмете Патологическая физиология
- #
- #
- #
- #
- #
- #
- #
26.01.20181.49 Mб51Лечки кратко (Не Кокорева).wiz
- #
- #
- #
- #
Эритроцитопатии. Наиболее часто встречается – наследственный семейный сфероцитоз (микросфероцитоз, болезнь Минковского – Шоффара, белковозависимая мембранопатия).
Заболевание наследуется аутосомно-доминантным путем. В основе его развития лежит дефект структуры мембраны эритроцитов, что приводит к изменению их формы с дискоидной на сферическую. Такие эритроциты не деформируются и при прохождении через узкие капилляры теряют часть мембранного вещества, уменьшаются в размерах, разрушаются. Их мембрана становится высоко проницаемой для ионов натрия и воды. На удаление натрия расходуется больше энергии (глюкозы, АТФ), чем в норме. В крови, где глюкозы достаточно, натриевый насос обеспечивает выведение избытка натрия. В межсинусовых пространствах селезёнки, где содержание глюкозы снижено, натрий не выводится, что приводит к осмотическому гемолизу эритроцитов. Основными клиническими проявлениями заболевания являются периодические гемолитические кризы, анемия, желтуха, спленомегалия, уробилинемия, уробилинурия, повышение температуры, трофические язвы голени в результате микротромбоза.
При этом содержание Нb и эритроцитов в крови уменьшается, развивается нормохромия, микросфероцитоз, ретикулоцитоз (10 % и более), снижается осмотическая резистентность эритроцитов. Во время гемолитических кризов наблюдается нейтрофильный лейкоцитоз.
К наследственно-обусловленным эритроцитопатиям (мембранопатиям) относятся также овалоцитоз (эллиптоцитоз), стоматоцитоз, акантоцитоз и другие ГА, получившие свое название от присущей им характерной формы эритроцитов (см. выше).
Ферментопатии (энзимопатии) объединяют группу ГА, которые проявляются недостаточностью активности ферментов эритроцитов, участвующих в процессе их энергетического обеспечения. В странах, прилегающих к Средиземному морю, Латинской Америки, Африки, Азии часто встречается анемия, вызванная дефицитом активности глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ) эритроцитов. Существуют две основные мутантные формы данного фермента. Одна из них (форма В) распространена среди европейцев, другая (форма А) – среди негритянского населения Африки. Заболевание передается по кодоминантному типу, сцеплено с Х-хромосомой. Ген, отвечающий за продуцирование Г-6-АДГ эритроцитов, располагается в Х-хромосоме рядом с геном цветного зрения и геном гемофилии. Лица, страдающие дефицитом Г-6-ФДГ эритроцитов, как и лица с серповидноклеточной анемией, реже погибают от тропической малярии, что обусловливает преимущественное распространение этой патологии в «малярийных» регионах. Для болезни характерно раннее проявление, нередко в период новорожденности. Она может сочетаться с гемофилией и дальтонизмом и клинически проявляется главным образом у мужчин. У женщин яркая клиника возможна только в случае наличия у них гомозиготности по данному гену.
При недостаточной активности Г-6-ФДГ в эритроцитах нарушается аэробное окисление глюкозы, что ослабляет процессы образования восстановленного НАДФ и восстановления глютатиона, необходимого для защиты Нb и мембраны эритроцитов от окислителей, в том числе и лекарственных веществ. При приеме обычных лечебных доз лекарств – окислителей (противомалярийных препаратов, сульфаниламидов, производных салициловой кислоты и др.) происходит окисление Нb, гем исчезает из его молекулы, выпадают в осадок цепи глобина в виде телец Гейнца. Эритроциты освобождаются от них в селезёнке. При этом утрачивается часть их мембранного вещества, в результате чего они подвергаются гемолизу, развивается гемолитический криз, прекращающийся после того, как все эритроциты с дефицитом Г-6-ФДГ разрушаются (феномен «самоограничения» гемолиза). Аналогичная картина наблюдается также при приеме с пищей конских бобов (фавизм – «багдадская весенняя лихорадка», распространена в Ираке в период цветения бобовых растений), иногда при вирусных инфекциях, гиповитаминозах Р, С, Е, отравлениях анилином, бензолом, фенилгидразином, в результате приема с пищей в больших количествах голубики, черники, вдыхания пыльцы трав, деревьев и т.д. (болезнь встречается в Беларуси).
Для гемолитических кризов характерны: высокая температура, головная боль, адинамия, гемоглобинурия, желтуха, гепатомегалия. Эти явления обусловливаются освобождающимися при повреждении эритроцитов медиаторами воспаления, в том числе и пирогенными цитокинами.
В картине крови отмечаются: Нb – 20-40 г/л, эритроциты до 1х1012/л, ретикулоцитоз, эритроциты с тельцами Гейнца, анизоцитоз, пойкилоцитоз, дегмациты, шизоциты, базофильная пунктация эритроцитов, нормобластоз, нейтрофильный лейкоцитоз со сдвигом влево (до миелоцитов).
Гемоглобинопатии (гемоглобинозы) возникают в результате наследственных нарушений синтеза глобина. Они могут быть качественные, обусловленные изменением первичной структуры Нb (серповидноклеточная анемия), и количественные, обусловленные нарушением скорости процесса синтеза одной из цепей глобина (талассемии). Большинство гемоглобинопатий наследуется аутосомно-доминантно. Данная патология встречаются главным образом в странах жаркого климата: в Центральной Африке, Азии, на Кубе. В некоторых районах Центральной Африки носительство гена серповидноклеточной анемии достигает 40-45 %. Гомозиготное носительство дает высокую детскую смертность.
Серповидноклеточная анемия (гемоглобинопатия S, дрепаноцитоз) – наиболее частая форма патологии, связанная с аномалией структуры Нb. Распространена она во многих тропических районах Африки, где малярия носит эндемический характер. Возникает эта патология, когда в ?— цепи Нb глютаминовая аминокислота заменяется на валин, что ведет к изменению физико-химических свойств молекулы гемоглобина (HbS). В восстановленном состоянии растворимость НbS резко снижается, молекулы агрегируют, и в результате образуется гель и кристаллы. Появляющиеся при этом полимеры представляют собой длинные нити, группирующиеся в так называемые тактоиды. Последние изменяют форму эритроцита, в результате чего формируются серповидные эритроциты (дрепаноциты), которые легко подвергаются гемолизу. В дрепаноцитах погибают малярийные плазмодии. Причина их гибели – снижение концентрации калия, возникающее в эритроците в состоянии дезоксигенации НbS, обусловленного повреждением плазматической мембраны и физическим повреждением паразитов агрегатами Нb.
Клинически заболевание проявляется в том случае, если содержание НbS в эритроцитах превышает 45 % или менее того, но при попадании больного в условия сниженного парциального давления кислорода (высокогорье, высотный полет и т.п.). При этом периодически возникают гемолитические, апластические, полиурические, никтурические, острые болевые, окклюзионные кризы. Их провоцируют гипоксия и ацидоз любого происхождения. Болевые приступы связаны с агрегацией дрепаноцитов в кровеносном русле, формированием микроэмболов, микротромбозом сосудов с развитием инфарктов различных органов, инсультов, «грудного синдрома» (окклюзия ветвей легочной артерии), ишемией и отслойкой сетчатки. Хроническая гипоксия и нарушение текучести крови приводят к гиперфункции миокарда и перегрузочной сердечной недостаточности. У больных отмечается вторичный иммунодефицит, повышенная восприимчивость к инфекциям, особенно в детском возрасте.
Для картины крови данного заболевания характерны анемия со значительным снижением числа эритроцитов и Нb, гипо- или нормохромия, анизоцитоз, пойкилоцитоз, базофильная пунктация эритроцитов, наличие дрепаноцитов, ретикулоцитоз, иногда нормобластоз, во время гемолитического криза – нейтрофильный лейкоцитоз со сдвигом влево, тромбоцитоз.
Талассемии (болезнь Кули, средиземноморская анемия) объединяют группу наследственных анемий, при которых наличие мутантного гена приводит к торможению синтеза цепей глобина, дефициту НbА.
Различают ?- и?-талассемию. Чаще встречается?-талассемия, при которой отсутствует или уменьшен синтез?-цепей глобина. В этом случае уменьшается количество НbА, в состав которого входят по две?- и?-цепи, а содержание НbА2(по две?- и?-цепи) и НbF (по две?- иA?- цепи) возрастает. Избыточно синтезирующиеся?-цепи образуют нестабильный Нb, возникают его преципитаты, содержащие их эритроциты удаляются клетками макрофагально-фагоцитарной системы. При этом повреждается мембрана эритроцитов, лишние?-цепи взаимодействуя сSH-группами этой мембраны, увеличивают ее проницаемость, что также способствует повышеннию гемолиза эритроцитов. Нарушается синтез гема и метаболизм железа.
У гомозигот развивается тяжелая гемолитическая анемия (большая талассемия, болезнь Кули), приводящая к высокой детской смертности на 1-м или 5-8-м году жизни. Характерен «монголоидный» тип лица, бледность и желтушность кожных покровов, язвы на нижних конечностях, спленомегалия, отставание в росте и развитии; рентгенологически у больных выявляется череп «ежика» (игольчатый периост теменных и лобных костей).
Гетерозиготы по генам талассемии отличаются повышенной устойчивостью к малярии (эритроциты с сокращенным сроком жизни ранее подвергаются фагоцитозу, при котором плазмодий гибнет). Эта форма ?-талассемии протекает значительно легче, чем другие формы.
При нарушении синтеза ?-цепей возникает?-талассемия. Гомозиготное носительство приводит к внутриутробной гибели плода, гетерозиготное – к гемолитической анемии различной тяжести.
В картине крови отмечаются гипохромная анемия (ЦП = 0,5 – 0,4), анизоцитоз, микроцитоз, пойкилоцитоз, гипохромия, большое количество мишеневидных эритроцитов (тороцитов), базофильная пунктуация эритроцитов; ретикулоцитоз (5-10 %), умеренный нейтрофильный лейкоцитоз со сдвигом влево, повышение уровеня сывороточного железа. Существует двойное гетерозиготное носительство аномальных алельных (структурных) и неалельных (структурных и регуляторных) генов, которое приводит к тяжелой гемолитической анемии. Например, аномальный НbЕ и?-талассемия,НbS/?-талассемия,НbН/?-талассемия и др. Близкородственные браки среди людей с высоким уровнем носительства аномальных гемоглобинов могут привести к увеличению числа гомозигот и двойных гетерозигот.
Распространение гемоглобинозов совпадает с так называемыми малярийными поясами Земли. Оказалось, что носители HbS и больные талассемией либо не болеют тропической малярией, либо переносят ее в легкой форме. Устойчивость больных гемоглобинозами к малярии объясняется тем, что возбудители ее являются внутриклеточными (внутриэритроцитарными) паразитами. Они потребляют большое количество кислорода, провоцируя тем самым ускоренный гемолиз эритроцитов, в процессе которого и сами погибают. Поскольку бессимптомное носительство HbS или малые формы талассемии не наносят организму серьезный вред, можно говорить о том, что одна менее тяжелая патология (легкие формы гемоглобинозов) становится защитным фактором по отношению к другому более тяжелому заболеванию (малярия).
Приобретенные гемолитические анемии возникают при появлении аутоантител к собственным эритроцитам организма (аутоиммунные); воздействии изоиммунных антител (переливание несовместимой крови, гемолитическая болезнь новорожденных); лекарственных веществ (сульфаниламиды и пр.); механическом повреждении эритроцитов (протезирование клапанов сердца, маршевая гемоглобинурия и пр.); вирусных инфекциях, действии химических и физических факторов (соли свинца, яды змей, ожоги, ультрафиолетовое облучение и пр.).
Гемолиз эритроцитов при данной форме анемии обусловливается метаболическими и структурными повреждениями их мембран, последующим повышением осмомолярности внутриклеточного содержимого, снижением способности эритроцитов к деформациям в синусах селезёнки, что способствует их разрушению.
В картине крови в первые часы развития анемии отмечается кратковременная «ложная» гиперхромия, затем развиваются нормохромная или гипохромная анемия, ретикулоцитоз, полихроматофилия, лейкоцитоз, увеличение в крови содержания непрямого билирубина.
