Молекулярные механизмы патогенеза атеросклероза
Атеросклероз
– хроническое прогрессирующее заболевание
крупных и средних эластических и
мышечно-эластических артерий. Атеросклероз
характеризуется пролиферативно-синтетическим
ответом ряда клеток сосудистой стенки
и крови – гладкомышечных макрофагов,
тромбоцитов, фибробластов на патологические
(качественно своеобразные или количественно
избыточные) ЛП, с формированием в интиме
фиброатером.
Причины
развития атеросклероза:
Гиперхолестеринемия;
Гиперлипидемия
ЛПОНП, ЛППП и ЛПНП (вызывают генетические
дефекты рецепторов, апобелков, СД,
гипотериоз, переедание).Изменение
нормальной структуры ЛПНП под действием
ПОЛ и гипергликемии. Избыток глюкозы
гликозилирует апобелки, повышенное
ПОЛ (при гипоксии, воспалении) повреждает
липиды и апобелки ЛП. Модифицированные
ЛПНП становятся чужеродными для
организма, атакуются антителами и
поглощаются макрофагами с участием
«скевенджер-рецепторов»
(рецепторов-мусорщиков);Повреждение
сосудистой стенки высоким артериальным
давлением (психоэмоциональные стрессы),
ПОЛ (гипоксия, курение (через СО),
воспаления), иммунными реакциями,
токсинами и другими ядовитыми веществами
(Pb,
Cd).
Повреждающие факторы разрыхляют и
истончают (до исчезновения) гликокаликс
энтероцитов, увеличивают межэндотелиальные
щели, что создает на поверхности
эндотелия зоны повышенной клейкости
и проницаемости;Принадлежность
к мужскому полу (гормональный статус).
Молекулярные
механизмы развития атеросклероза
Развитие
атеросклероза проходит в 6 стадий:
Стадия
измененного эндотелия.
На поверхности поврежденного эндотелия
скапливаются тромбоциты и моноциты.
Модифицированные ЛПНП проникают под
поврежденный эндотелий сосудов. За
ними направляются моноциты (в ткани
они макрофаги) и захватывают ЛП через
скевенджер-рецепторы. Этот процесс не
ингибируется избытком ХС, поэтому
макрофаги перегружаются ХС и превращаются
в «пенистые клетки».
Отдельные «пенистые клетки» есть у
новорожденных.Стадия
жировых полосок.
При увеличении количества «пенистых
клеток» они образуют липидные
полоски.
«Пенистые» клетки адсорбируют все
остальные липиды без разбора. Поврежденный
эндотелий, активированные макрофаги,
тромбоциты выделяют БАВ, которые
стимулируют пролиферацию ГМК и миграцию
их в очаг повреждения.Стадия
переходная.
Активированные ГМК синтезируют коллаген
и эластин, что приводит к прорастанию
бляшки фиброзной тканью. Клетки под
фиброзной оболочкой некротизируются,
а ХС начинает откладываться в межклеточном
пространстве. Может происходить разрыв
эндотелия сосудов.Стадия
атеромы. ХС
межклеточного пространства формирует
в центре бляшки липидную каплю –
атерому,
которая через разрушенный эндотелий
выступает в просвет сосуда.Стадия
фиброатеромы.
Атерома пропитываясь солями кальция,
белками, ГАГ и приобретает плотную
фиброзную крышку. Атерома становиться
фиброатеромой.Стадия
осложнения фиброатеромы.
Фиброатерома не стабильна, она может
надрываться и изъявляться, что приводит
к обострению атеросклероза.
Осложнения.
Поврежденный эндотелий прекращает
синтез PGI2,
который в норме ингибирует тромбоциты.
Тромбоциты активируются и секретируют
тромбоксан ТХА2
и тромбоцитарный фактор роста (пептид).
Тромбоцитарный фактор роста привлекает
в бляшку клетки крови, ГМК, что способствует
росту бляшки и развитию очага воспаления.
ТХА2 →
агрегацию
тромбоцитов →
образование тромбов → закупорка сосудов
→ ишемия тканей → некроз тканей →
изъявления стенок сосудов → кровотечения,
аневризмы. Оторвавшиеся тромбы → эмболии
сосудов.
Чаще
всего атеросклероз развивается в
коронарных, мозговых, почечных артериях,
артериях нижних конечностей и в аорте.
Атеросклероз коронарных артерий
проявляется ИБС, мозговых – ИБ мозга,
почек – вазоренальной артериальной
гипертензией. Спазм или тромбоз коронарных
сосудов ведет к инфаркту миокарда,
эмболия сонных артерий ведет к развитию
инсультов.
Смертность
от последствий атеросклероза (инфаркт
миокарда, инсульт) лидирует в общей
структуре смертности населения.
Биохимические
основы лечения атеросклероза
Лечение
гиперхолестеролемии, как правило,
комплексное.
I
Диета. Необходимо
употреблять:
продукты
гипокалорийные, гипохолестериные, с
низким содержанием легкоусвояемых
углеводов (растительная пища). Поступление
ХС с пищей не должно превышать 0,3 мг/сут;полиеновые
ЖК семейства ω-3 (морепродукты). Из них
синтезируются простагландины,
подавляющие тромбообразование и
замедляют развитие атеросклеротической
бляшки. Ненасыщенные ЖК также ускоряют
выведение ХС из организма (механизм
не ясен);витамины
С, Е, А и другие антиоксиданты ингибирующие
ПОЛ и поддерживающие нормальную
структуру ЛПНП и их метаболизм.
Липримал
дает самый сильный эффект
II.
«Размыкание» цикла энтерогепатической
циркуляции жёлчных кислот. Лекарства
типа холестирамина, холестипол
(полимеры) адсорбируют в кишечнике
жёлчные кислоты, выделяются с фекалиями
и таким образом уменьшают возврат
жёлчных кислот в печень. В печени
увеличивается захват ХС из крови для
синтеза новых жёлчных кислот.
III.
Ингибирование синтеза ХС.
Наиболее эффективные препараты для
лечения атеросклероза — ингибиторы
ГМГ-КоА-редуктазы, например антибиотик
мевакор.
Такие препараты могут почти полностью
подавить синтез ХС в организме, нормализуя
уровень ХС.
IV.
Активация катаболизма ЛП.
Лекарственные препараты — фибраты
(клофибрат, фенофибрат) активируют ЛПЛ
и ускоряют катаболизм ЛПОНП. Эти препараты
также активируют окисление ЖК в печени,
уменьшая тем самым синтез ТГ и ЭХС и,
как следствие, секрецию ЛПОНП печенью.
Для
эффективного лечения атеросклероза
применяют, как правило, комбинированное
воздействие нескольких лекарственных
препаратов.
|

Берсенёв Алексей Вячеславович диссертация
По
современным представлениям атеросклероз
– это хроническая системная воспалительная
реакция организма, развивающаяся на
фоне дислипидемии и сопровождающаяся
образованием одиночных или множественных
очагов липидных отложений (атероматозных
бляшек) на внутренней поверхности
сосудов [134].
Полагают,
что именно системная воспалительная
реакция способствует развитию дислипидемии
(ДЛП) и запускает процесс атерогенеза
[33,53]. В свою очередь алиментарные и
наследственные ДЛП также индуцируют
проявления синдрома системного
воспалительного ответа и усугубляют
тяжесть атеросклеротического поражения
сосудов в организме [12,13,18,22,43].
В
присутствии провоспалительных факторов,
таких как окисленные ЛП (в особенности
низкой и очень низкой плотности)
[16,18,43], инфекционные агенты [33,53] и
различные неспецифические стресс-факторы,
в организме активируется
макрофагально-моноцитарная система и
усиливается выработка провоспалительных
цитокинов (интерлейкинов: IL-1, 6, фактора
некроза опухоли: TNF-α и др.). Эти цитокины,
с одной стороны, вызывают в сосудистом
эндотелии экспрессию молекул адгезии
– ICAM-1, ICAM-2 (intracellular adhesion molecules), VCAM-1
(vascular cell adhesion molecules), селектины и др. и
нарушают структуру эндотелиальной
выстилки сосудов, а с другой вызывают
экспрессию в гепатоцитах генов,
ответственных за синтез в печени
острофазных белков. Участие печени в
острофазном процессе и, следовательно,
в инициации и модуляции системной
воспалительной реакции организма
[66,134], смещает в ней баланс биохимических
механизмов, что вызывает прежде всего,
нарушения липидного обмена, поскольку
печень играет центральную роль в
регуляции этого вида обмена в организме.
Длительно поддерживаемая в организме
активация макрофагально-моноцитарной
системы из адаптивной постепенно
превращается в повреждающую, при которой
не только нарушается регуляция печенью
липидного обмена, но и создаются условия
для прогрессирования ДЛП и атеросклероза,
а также для развития их осложнений
[12,13,22].
При
ДЛП и атеросклерозе клетками-мишенями
(при системной воспалительной реакции)
являются, прежде всего, клетки печени
– гепатоциты, купферовские клетки,
эндотелиоциты, а также эндотелиальная
выстилка сосудов [16,17], изменения в
которых развиваются параллельно,
постепенно прогрессируют, ведут к
формированию хронического гепатита
[17], а также к типичному повреждению
сосудистой стенки атеросклеротическим
процессом.
Соседние файлы в предмете Биохимия
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Одним из самых ярких и клинически значимых нарушений обмена липопротеинов является атеросклероз.
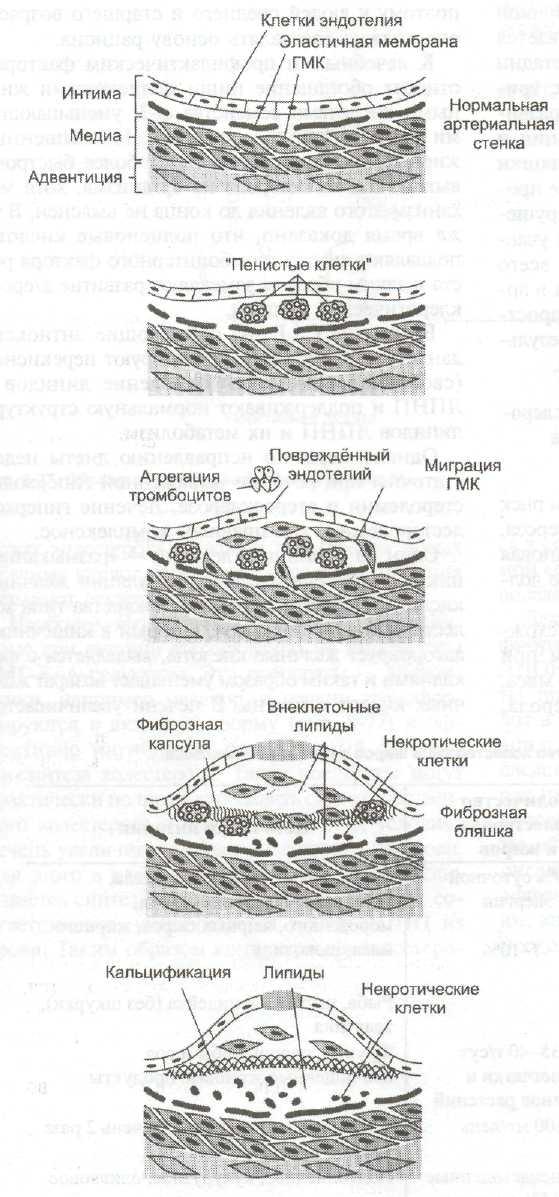
Атеросклероз
Атеросклероз – это отложение холестерина и его эфиров в соединительной ткани стенок артерий, в которых выражена механическая нагрузка на стенку (по убыванию воздействия): абдоминальная аорта, коронарная артерия, подколенная артерия, бедренная артерия, тибиальная артерия, грудная аорта, дуга грудной аорты, сонные артерии.
Стадии атеросклероза
Морфологически выделяют четыре стадии атеросклероза. Первая и вторая стадии распространены широко и при правильном питании являются обратимыми, 3 и 4 стадии уже имеют клиническое значение и необратимы.
1 стадия – повреждение эндотелия
Это «долипидная» стадия, обнаруживается даже у годовалых детей. Изменения этой стадии неспецифичны и ее могут вызывать: дислипопротеинемия, гипертензия, повышение вязкости крови, курение, вирусные и бактериальные инфекции, свинец, кадмий и т.п.
На этой стадии в эндотелии создаются зоны повышенной проницаемости и клейкости. Внешне это проявляется в разрыхлении и истончении (вплоть до исчезновения) защитного гликокаликса на поверхности эндотелиоцитов, расширении межэндотелиальных щелей. Это приводит к усилению выхода моноцитов и липопротеинов (ЛПНП и ЛПОНП) в интиму.
2 стадия – стадия начальных изменений
Отмечается у большинства детей и молодых людей.
Поврежденный эндотелий и активированные тромбоциты вырабатывают медиаторы воспаления, факторы роста, эндогенные окислители. В результате через поврежденный эндотелий в интиму сосудов еще более активно проникают моноциты и способствуют развитию воспаления. При этом ЛПНП, попавшие под интиму, начинают изменяться (модифицироваться), т.е. подвергаются окислению, гликозилированию, ацетилированию.
Моноциты, преобразуясь в макрофаги, активно поглощают измененные липопротеины при участии «мусорных» рецепторов (scavenger [‘skævɪnʤə] receptors). Таким образом, поглощение модифицированных ЛПНП макрофагами идет без участия апоВ-100-рецепторов, а, значит, нерегулируемо.
При поглощении модифицированных липопротеинов макрофаги активируются, выделяют цитокины и разнообразные факторы роста, которые стимулируют деление гладкомышечных клеток, синтез межклеточного вещества, и играют роль в развитии атеросклеротической бляшки.
Модификация липопротеинов в зоне воспаления является непосредственной биохимической причиной атеросклероза.
Окисление ЛПНП нарастает при недостаточной активности антиоксидантных систем – гиповитаминозах Е и С, нехватке металлов (железо, селен, медь, цинк), входящих в состав антиоксидантных ферментов каталазы, пероксидазы, супероксиддисмутазы.
Гликозилирование белков ЛПНП ускоряется при сахарном диабете или при других хронических гипергликемиях. Такие модифицированные липопротеины теряют способность связываться с апоВ-100-рецептором и проникать в клетки-мишени и, в результате, накапливаются в крови и в интиме сосудов.
Под действием факторов роста гладкомышечные клетки медии мигрируют в интиму и начинают пролиферировать, превращаясь в макрофагоподобные клетки. Они также накапливают модифицированные ЛПНП.
Накопление липидов в макрофагах быстро исчерпывает невысокие возможности клеток по утилизации свободного и этерифицированного ХС. Они переполняются стероидами и превращаются в пенистые клетки. Внешне на эндотелии появляются липидные пятна и полоски.
Процесс развития атеросклероза (в динамике слева-направо)
3 стадия – стадия поздних изменений
Продолжают развертываться и приобретают масштабность события, начавшиеся на второй стадии.
Внешне проявляется как выступание поверхности в просвет сосуда. Стадия дополнительно характеризуется следующими особенностями:
- увеличение количества коллагена, эластина и гликозаминогликанов, т.е. накопление межклеточного вещества,
- пролиферация и гибель пенистых клеток (апоптоз),
- накопление в межклеточном пространстве свободного ХС и этерифицированного ХС,
- инкапсулирование холестерола и формирование фиброзной бляшки.
4 стадия – стадия осложнений
На этой стадии происходят:
- кальцификация бляшки и ее изъязвление, приводящее к эмболии сосудов,
- тромбоз из-за адгезии и активации тромбоцитов,
- разрыв сосуда.
Основы лечения
В лечении атеросклероза обязательно должны быть две составляющие: диета и медикаменты. Целью лечения является снижение концентрации общего ХС плазмы, ХС ЛПНП и ЛПОНП, повышение концентрации ЛПВП.
Диета
1. Обеспечение организма витаминами: аскорбиновой кислотой, пантотеновой (коэнзим А) и никотиновой (НАДФ) кислотами, что способствует превращению холестерола печени в желчные кислоты (синтез желчных кислот). Для снижения окислительной модификации ЛПНП необходим витамин Е.
2. Снижение калорийности пищи за счет углеводов и жиров. Жиры пищи должны включать равные доли насыщенных, мононенасыщенных и полиненасыщенных жирных кислот. Доля жидких жиров, содержащих полиненасыщенные жирные кислоты (ПНЖК), должна быть около 30% от всех жиров, но не меньше 15 г/сут. Роль ПНЖК в лечении гиперхолестеролемии и атеросклероза сводится к:
- ограничению всасывания ХС в тонком кишечнике,
- активации синтеза фосфатидилхолина, что снижает вязкость желчи и облегчает ее отток в кишечник,
- усилению желчеотделения,
- снижению синтеза ЛПНП в печени и секреции их в кровь,
- увеличению синтеза ЛПВП и концентрации их в крови, что способствует удалению холестерина из тканей в печень.
3. Обеспечение организма чистой водой до физиологических норм (1,0-1.5 л/сут), что препятствует сгущению желчи.
4. Потребление высоких количеств овощей, содержащих целлюлозу (капуста, морковь, свекла) для усиления перистальтики кишечника, стимуляции желчеотделения и снижения всасывания ХС.
5. Умеренная физическая нагрузка – способствует синтезу ЛПВП и, значит, оттоку холестерина от тканей в печень.
Медикаменты
1. Препараты ω6- и ω3-жирных кислот (Линетол, Эссенциале, Омеганол и т.п.) повышают концентрацию ЛПВП в плазме, ускоряют отток ЛПНП в печень, стимулируют желчеотделение.
2. Подавление всасывания ХС в желудочно-кишечном тракте – анионообменные смолы (Холестирамин, Холестид, Questran).
3. Высокие дозы никотиновой кислоты подавляют мобилизацию жирных кислот из депо и снижают синтез ЛПОНП в печени, а, следовательно, и образование из них ЛПНП в крови.
4. Фибраты (клофибрат и т.п.) увеличивают активность липопротеинлипазы, ускоряют катаболизм ЛПОНП и хиломикронов, что повышает переход холестерола из них в ЛПВП и его эвакуацию в печень.
5. Статины (ловастатин, флувастатин) ингибируют ГМГ-SКоА-редуктазу, что снижает в 2 раза синтез ХС в печени и ускоряют его отток из ЛПВП в гепатоциты.
Предложены и совсем радикальные способы:
6. Подавление функции энтероцитов с помощью антибиотика неомицина, что снижает всасывание жиров.
7. Хирургическое удаление подвздошной кишки и прекращение реабсорбции желчных кислот.
Атеросклероз
– хроническое прогрессирующее заболевание
крупных и средних эластических и
мышечно-эластических артерий. Атеросклероз
характеризуется пролиферативно-синтетическим
ответом ряда клеток сосудистой стенки
и крови – гладкомышечных макрофагов,
тромбоцитов, фибробластов на патологические
(качественно своеобразные или количественно
избыточные) ЛП, с формированием в интиме
фиброатером.
Причины
развития атеросклероза:
Гиперхолестеринемия;
Гиперлипидемия
ЛПОНП, ЛППП и ЛПНП (вызывают генетические
дефекты рецепторов, апобелков, СД,
гипотериоз, переедание).Изменение
нормальной структуры ЛПНП под действием
ПОЛ и гипергликемии. Избыток глюкозы
гликозилирует апобелки, повышенное
ПОЛ (при гипоксии, воспалении) повреждает
липиды и апобелки ЛП. Модифицированные
ЛПНП становятся чужеродными для
организма, атакуются антителами и
поглощаются макрофагами с участием
«скевенджер-рецепторов»
(рецепторов-мусорщиков);Повреждение
сосудистой стенки высоким артериальным
давлением (психоэмоциональные стрессы),
ПОЛ (гипоксия, курение (через СО),
воспаления), иммунными реакциями,
токсинами и другими ядовитыми веществами
(Pb,
Cd).
Повреждающие факторы разрыхляют и
истончают (до исчезновения) гликокаликс
энтероцитов, увеличивают межэндотелиальные
щели, что создает на поверхности
эндотелия зоны повышенной клейкости
и проницаемости;Принадлежность
к мужскому полу (гормональный статус).
Молекулярные
механизмы развития атеросклероза
Развитие
атеросклероза проходит в 6 стадий:
Стадия
измененного эндотелия.
На поверхности поврежденного эндотелия
скапливаются тромбоциты и моноциты.
Модифицированные ЛПНП проникают под
поврежденный эндотелий сосудов. За
ними направляются моноциты (в ткани
они макрофаги) и захватывают ЛП через
скевенджер-рецепторы. Этот процесс не
ингибируется избытком ХС, поэтому
макрофаги перегружаются ХС и превращаются
в «пенистые клетки».
Отдельные «пенистые клетки» есть у
новорожденных.Стадия
жировых полосок.
При увеличении количества «пенистых
клеток» они образуют липидные
полоски.
«Пенистые» клетки адсорбируют все
остальные липиды без разбора. Поврежденный
эндотелий, активированные макрофаги,
тромбоциты выделяют БАВ, которые
стимулируют пролиферацию ГМК и миграцию
их в очаг повреждения.Стадия
переходная.
Активированные ГМК синтезируют коллаген
и эластин, что приводит к прорастанию
бляшки фиброзной тканью. Клетки под
фиброзной оболочкой некротизируются,
а ХС начинает откладываться в межклеточном
пространстве. Может происходить разрыв
эндотелия сосудов.Стадия
атеромы. ХС
межклеточного пространства формирует
в центре бляшки липидную каплю –
атерому,
которая через разрушенный эндотелий
выступает в просвет сосуда.Стадия
фиброатеромы.
Атерома пропитываясь солями кальция,
белками, ГАГ и приобретает плотную
фиброзную крышку. Атерома становиться
фиброатеромой.Стадия
осложнения фиброатеромы.
Фиброатерома не стабильна, она может
надрываться и изъявляться, что приводит
к обострению атеросклероза.
Осложнения.
Поврежденный эндотелий прекращает
синтез PGI2,
который в норме ингибирует тромбоциты.
Тромбоциты активируются и секретируют
тромбоксан ТХА2
и тромбоцитарный фактор роста (пептид).
Тромбоцитарный фактор роста привлекает
в бляшку клетки крови, ГМК, что способствует
росту бляшки и развитию очага воспаления.
ТХА2 →
агрегацию
тромбоцитов →
образование тромбов → закупорка сосудов
→ ишемия тканей → некроз тканей →
изъявления стенок сосудов → кровотечения,
аневризмы. Оторвавшиеся тромбы → эмболии
сосудов.
Чаще
всего атеросклероз развивается в
коронарных, мозговых, почечных артериях,
артериях нижних конечностей и в аорте.
Атеросклероз коронарных артерий
проявляется ИБС, мозговых – ИБ мозга,
почек – вазоренальной артериальной
гипертензией. Спазм или тромбоз коронарных
сосудов ведет к инфаркту миокарда,
эмболия сонных артерий ведет к развитию
инсультов.
Смертность
от последствий атеросклероза (инфаркт
миокарда, инсульт) лидирует в общей
структуре смертности населения.
Биохимические
основы лечения атеросклероза
Лечение
гиперхолестеролемии, как правило,
комплексное.
I
Диета. Необходимо
употреблять:
продукты
гипокалорийные, гипохолестериные, с
низким содержанием легкоусвояемых
углеводов (растительная пища). Поступление
ХС с пищей не должно превышать 0,3 мг/сут;полиеновые
ЖК семейства ω-3 (морепродукты). Из них
синтезируются простагландины,
подавляющие тромбообразование и
замедляют развитие атеросклеротической
бляшки. Ненасыщенные ЖК также ускоряют
выведение ХС из организма (механизм
не ясен);витамины
С, Е, А и другие антиоксиданты ингибирующие
ПОЛ и поддерживающие нормальную
структуру ЛПНП и их метаболизм.
Липримал
дает самый сильный эффект
II.
«Размыкание» цикла энтерогепатической
циркуляции жёлчных кислот. Лекарства
типа холестирамина, холестипол
(полимеры) адсорбируют в кишечнике
жёлчные кислоты, выделяются с фекалиями
и таким образом уменьшают возврат
жёлчных кислот в печень. В печени
увеличивается захват ХС из крови для
синтеза новых жёлчных кислот.
III.
Ингибирование синтеза ХС.
Наиболее эффективные препараты для
лечения атеросклероза — ингибиторы
ГМГ-КоА-редуктазы, например антибиотик
мевакор.
Такие препараты могут почти полностью
подавить синтез ХС в организме, нормализуя
уровень ХС.
IV.
Активация катаболизма ЛП.
Лекарственные препараты — фибраты
(клофибрат, фенофибрат) активируют ЛПЛ
и ускоряют катаболизм ЛПОНП. Эти препараты
также активируют окисление ЖК в печени,
уменьшая тем самым синтез ТГ и ЭХС и,
как следствие, секрецию ЛПОНП печенью.
Для
эффективного лечения атеросклероза
применяют, как правило, комбинированное
воздействие нескольких лекарственных
препаратов.
|
Берсенёв Алексей Вячеславович диссертация
По
современным представлениям атеросклероз
– это хроническая системная воспалительная
реакция организма, развивающаяся на
фоне дислипидемии и сопровождающаяся
образованием одиночных или множественных
очагов липидных отложений (атероматозных
бляшек) на внутренней поверхности
сосудов [134].
Полагают,
что именно системная воспалительная
реакция способствует развитию дислипидемии
(ДЛП) и запускает процесс атерогенеза
[33,53]. В свою очередь алиментарные и
наследственные ДЛП также индуцируют
проявления синдрома системного
воспалительного ответа и усугубляют
тяжесть атеросклеротического поражения
сосудов в организме [12,13,18,22,43].
В
присутствии провоспалительных факторов,
таких как окисленные ЛП (в особенности
низкой и очень низкой плотности)
[16,18,43], инфекционные агенты [33,53] и
различные неспецифические стресс-факторы,
в организме активируется
макрофагально-моноцитарная система и
усиливается выработка провоспалительных
цитокинов (интерлейкинов: IL-1, 6, фактора
некроза опухоли: TNF-α и др.). Эти цитокины,
с одной стороны, вызывают в сосудистом
эндотелии экспрессию молекул адгезии
– ICAM-1, ICAM-2 (intracellular adhesion molecules), VCAM-1
(vascular cell adhesion molecules), селектины и др. и
нарушают структуру эндотелиальной
выстилки сосудов, а с другой вызывают
экспрессию в гепатоцитах генов,
ответственных за синтез в печени
острофазных белков. Участие печени в
острофазном процессе и, следовательно,
в инициации и модуляции системной
воспалительной реакции организма
[66,134], смещает в ней баланс биохимических
механизмов, что вызывает прежде всего,
нарушения липидного обмена, поскольку
печень играет центральную роль в
регуляции этого вида обмена в организме.
Длительно поддерживаемая в организме
активация макрофагально-моноцитарной
системы из адаптивной постепенно
превращается в повреждающую, при которой
не только нарушается регуляция печенью
липидного обмена, но и создаются условия
для прогрессирования ДЛП и атеросклероза,
а также для развития их осложнений
[12,13,22].
При
ДЛП и атеросклерозе клетками-мишенями
(при системной воспалительной реакции)
являются, прежде всего, клетки печени
– гепатоциты, купферовские клетки,
эндотелиоциты, а также эндотелиальная
выстилка сосудов [16,17], изменения в
которых развиваются параллельно,
постепенно прогрессируют, ведут к
формированию хронического гепатита
[17], а также к типичному повреждению
сосудистой стенки атеросклеротическим
процессом.
Соседние файлы в предмете Химия
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
27.03.2016101.38 Кб214химия1.xls
