Дэб тест при анемии
Анемия Фанкони (АФ) — редкое наследственное аутосомно рецессивное заболевание, характеризующееся недостатком кроветворе ния, аномалиями развития и склонностью к развитию опухолей вследствие нестабильно сти генома.
Частота встречаемости 1:350 000 новорож денных, частота носительства по разным ис точникам 1:181—300 [1, 2].
В настоящее время известно 19 генов, связан ных с развитием АФ, каждый из которых от вечает за синтез определенного протеина, так или иначе участвующего в процессе репара ции ДНК [3, 4, 5].
КЛИНИЧЕСКИЙ ПРИМЕР
В январе 2017 г. в гематологическое отделение МДГКБ поступил мальчик М., 1 год 3 мес.
Анамнез. Ребенок от 1 беременности, род ственный брак (родители двоюродные сибсы). Роды 1 на 41 нед, самостоятельные, воды про зрачные.
Масса 2800 г, рост 49 см.
По ЭхоКГ открытый артериальный проток. Выписан на 5 сут в удовлетворительном состо янии, ребенок находился на грудном вскарм ливании по требованию.
В возрасте 2 мес жизни (с 02.12 по 18.12.2015) находился на лечении в ДГКБ № 9 с диагнозом «галактоземия? Холестатический синдром. Вторичный геморрагический синдром. Суб арахноидальное кровоизлияние. Постгемор рагическая анемия тяжелой степени. Дефи цит веса». В отделении у ребенка отмечалась двухростковая цитопения (эритропения, анемия, тромбоцитопения), гипокоагуляция, гипербилирубинемия. Проводилась терапия свежезамороженной плазмой № 3, двукратно введение эритроцитарной массы, гемостати ческая терапия викасолом и дициноном. Ге моррагический синдром купирован.
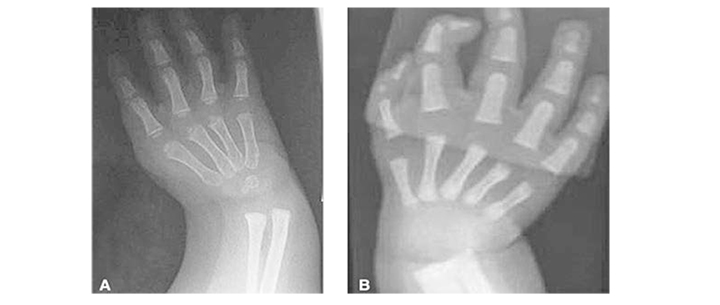
Настоящая госпитализация в гематологиче ское отделение МДГКБ в связи с появлением фебрильной лихорадки и продуктивного кашля на фоне агранулоцитоза и тромбоцитопении. При поступлении обращали на себя внимание задержка физического и интеллектуального развития, множественные стигмы дисэмбрио генеза, в том числе гипоплазия дистальной фаланги большого пальца левой руки; блед ность кожных покровов, умеренный гемор рагический синдром в виде петехиальных кровоизлияний и экхимозов на коже лица, конечностей.
В общем анализе крови трехростковая ци топения: эритроциты 2,16×1012/л, Нв 74 г/л, MCV 101 фл, MCH 34 пг, тромбоциты 22×109/л, лейкоциты 2,3×109/л, абсолютное число нейтрофилов 0,299×109/л.
В биохимическом анализе крови: умерен ное увеличение ЩФ (421 при Н до 345 Ед/л), ЛДГ (545 при Н до 430 Ед/л), АСТ (105 при Н до 50 Ед/л), АЛТ (74 при Н до 45 Ед/л), ферритина (433 при Н до 60 мкг/л).
При исследовании клеточного иммунитета небольшое увеличение иммунорегулятор ного индекса.
Коагулограмма — нормокоагуляция. ВИЧ, HCV HBsAg отрицательно.
В миелограмме выраженное снижение кле ток эритроцитарного ряда (0,8% при Н 14,5—26,5), сужение нейтрофильного ростка с «обрывом» на уровне миелоцитов, отсутствие мегакариоцитов.
ДЭБтест: положительный (исследовано 30 метафаз, выявлено 144 аберрации; карио тип 46 XY).
ТЕСТ НА ЛОМКОСТЬ ХРОМОСОМ
«Золотым стандартом» скрининга для выявле ния АФ был и остается тест с диэпоксибутаном (ДЭБ) и его вариант с митомицином С (MMC) [5, 6, 7].
Суть метода: после обработки лимфоцитов или фибробластов алкилирующим веществом (в нелетальной для клеток концентрации) определяют частоту и спектр спонтанных и индуцированных in vitro хромосомных абер раций.
Тест не имеет 100% специфичности, лишь очерчивая дифференциальный круг, в кото рый входит АФ, синдром Ниймегена, синдром Робертса, Warsaw breakage syndrome, синдром Блюма, врожденный дискератоз и некоторые другие синдромы. Поэтому в соответствии с рекомендациями по диагностике АФ страте гия генетического скрининга включает так же целевой скрининг мутаций для некоторых популяционных групп, выявление наиболее частых делеций гена FANCA методом MLPA, исследование известных генов АФ методом высокопроизводительного секвенирования. Таким образом, на основании анамнеза, клиниколабораторных данных ребенку был выставлен диагноз «анемия Фанкони».
Ребенку проводилась комплексная терапия, включающая заместительную терапию эри троцитарной взвесью и тромбоконцентра том, антибактериальная, противогрибковая и симптоматическая терапия, на фоне которой анемия, геморрагический синдром, агрануло цитоз, симптомы интоксикации, лихорадки были купированы. Ребенок выписан для даль нейшего амбулаторного наблюдения.
В настоящее время единственным радикаль ным способом лечения АФ является трансплан тация костного мозга от HLAсовместимого донора.
- Glanz A., Fraser F. C. Spectrum of anomalies in Fanconi anemia. J. Med. Genet. 19: 412416, 1982.
- Grompe M., D’Andrea A. Fanconi anemia and DNA repair. Hum. Molec. Genet. 10: 22532259, 2001.
- Auerbach A.D., Rogatko A., SchroederKurth T.M. International Fanconi Anemia Registry: relation of clinical symptoms to diepoxybutane sensitivity. Blood 73: 391396, 1989.
- Brunetti P., Neuci G.G., Vaccaro R., Puxeddu A., Migliorini E. Fanconi’s anaemia. (Letter). Lancet 288: 11941195, 1966. Note: Originally Volume II.
- Joenje H., Patel K.J. The emerging genetic and molecular basis of Fanconi anaemia. Nature Rev. Genet. 2: 446457, 2001.
- Alter B.P. Fanconi’s anaemia and its variability. Brit. J. Haemat. 85: 914, 1993.
- Генетическая диагностика анемии Фанкони. Обзор литературы. Панферова А.В., Тимофеева Н.М., Ольшан ская Ю.В. Онкогематология 3.2016 том 11. DOI 10.17650/1818834620161137685

Анемия Фанкони – это генетическое заболевание, которое передается по аутосомно-рецессивному типу и характеризуется нарушением кроветворения, формированием злокачественных новообразований, пороками развития, ломкостью хромосом. Проявляется частыми кровотечениями, кровоподтеками на коже, вялостью, бледностью, склонностью к инфекциям. Диагностика проводится лабораторными методами, назначаются цитогенетическое, молекулярно-генетическое и клиническое исследования крови, миелограмма. Основные способы лечения – пересадка костного мозга, медикаментозное поддержание кроветворения, переливание крови.
Общие сведения
Синонимичные названия анемии Фанкони – врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Заболевание названо по фамилии швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов у трех братьев. Анемия Фанкони является редкой генетической болезнью, наследуется согласно аутосомно-рецессивному принципу. Эпидемиологические показатели низкие – 1 больной ребенок на 350 тысяч новорожденных. Распространенность одинакова среди представителей женского и мужского пола, выше в сообществах с разрешенными близкородственными браками, например, у некоторых южноафриканских народов.

Анемия Фанкони
Причины
Заболевание является наследственным, развивается при передаче дефектного гена от родителей к ребенку. Выявлено 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена находится в X-хромосоме (сцепленной с полом). Все эти гены отвечают за производство определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что и отец, и мать должны быть носителями патологической генетической информации. При этом сами они, как правило, здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, получивших от каждого из родителей один и тот же измененный ген. В крайне редких случаях анемия провоцируется передачей дефектной Х-сцепленной хромосомы. Женщины могут быть носительницами мутации, заболевание проявляется только у мальчиков. Риск развития патологии у сына при наличии у матери мутированного гена – 50%.
Патогенез
В норме в клетках организма существуют специальные ферментные системы, которые исправляют разрывы молекул ДНК, поврежденных в процессе биосинтеза или воздействия химических, физических реагентов. При анемии Фанкони обнаруживается генетический дефект в кластере белков, ответственных за репарацию ДНК, что приводит к повышенной ломкости хромосом. В итоге у пациентов развиваются нарушения функций костного мозга – неоплазии и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью миелоидного ростка крови, провоцирующей накопление измененных белых клеток, подавляющих рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии в результате дисплазии костного мозга резко угнетается рост и созревание всех трех видов клеток крови.
Симптомы анемии Фанкони
Более чем у половины пациентов наблюдаются врожденные аномалии развития внутренних органов и скелета. Костные деформации проявляются специфическим внешним видом: больные низкорослые, с уменьшенным размером головы, отсутствием или заметным укорочением большого пальца на руках, недоразвитием лучевой кости, врожденным вывихом бедра и/или наличием шейного ребра, косолапостью, недоразвитым подбородком («птичьим лицом»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, глазным дрожанием, глухотой, умственной отсталостью. Больные зачастую имеют незрелые половые органы, у них отсутствует одно или оба яичка. К распространенным аномалиям строения органов относятся пороки мочевыделительной системы: удвоение мочеточников или лоханки, подковообразные почки, почечные кисты, смещенное наружное отверстие уретры (гипоспадия). Врожденные пороки сердца включают атрезию трехстворчатого клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Ключевые симптомы связаны с постепенным нарастанием нарушений в работе костного мозга. Чаще они дебютируют в детском возрасте (в 5-10 лет). Из-за снижения количества тромбоцитов развивается повышенная кровоточивость: при ранениях кровь долго не сворачивается, легко возникают носовые кровотечения, выделения при менструациях обильны, на теле обнаруживается много «беспричинных» кровоподтеков. Уменьшение числа эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружениями, обмороками, бледностью кожи, учащенным сердцебиением и одышкой. Недостаток лейкоцитов способствует ухудшению сопротивляемости инфекциям. Впоследствии формируется лейкоз, миелодиспластический синдром, онкологические болезни.
Осложнения
Наиболее распространенным осложнением считаются частые инфекционные заболевания. У пациентов развивается ОРВИ, ангина, ринит, бронхит, грипп, тиф, герпес. Рецидивирующий характер болезней и их тяжелое течение приводят к деструкции органов, сопровождаются риском сепсиса. Другим осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли органов шеи и головы, половых органов. Рак у таких больных тяжело поддается лечению из-за повышенной ломкости и сниженной репарации ДНК. Это явление ограничивает применение лучевой терапии, цитотоксических препаратов. Нарушение свертываемости становится причиной больших кровопотерь.
Диагностика
Обследование больных проводят онкологи, гематологи, педиатры, врачи-генетики. Диагностика начинается с анализа анамнестических данных и жалоб. Врач выясняет, имеется ли данное наследственное заболевание у близких родственников, уточняет время появления первых признаков болезни, ранние обращения к врачам. При осмотре оценивает общее состояние пациента, выявляет наличие аномалий развития, гиперпигментированных пятен, кровотечений, кровоподтеков. В большинстве случаев не составляет труда обнаружить типичные деформации костей, недоразвитие глаз. Для подтверждения диагноза и различения анемии Фанкони с приобретенной анапластической анемией проводится ряд лабораторных исследований:
- Клинический анализ крови. Характерны изменения клеточного состава крови. На ранних этапах нарушения кроветворения диагностируется тромбоцитопения и лейкопения, на более поздних – панцитопения (резкое снижение объема эритроцитов, лейкоцитов и тромбоцитов). Возможен умеренный гемолиз без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ увеличено до 60-80 мм/ч.
- Цитогенетическое исследование клеток. Выполняется проба с диэпоксибутаном, митомицином C, указывающая на частоту и спектр хромосомных аберраций. В пользу генетической анемии рассматриваются показатели ДЭБ-теста более 45%, пограничный уровень – 11-45% (процент клеток с хромосомными разрывами).
- Молекулярно-генетический анализ клеток. Исследуются гены, мутации в которых могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% – в аллели FANCC, в 10% – в генах FANCG. Частота мутаций в других парах – 0,2-3%.
- Миелограмма. По данным исследования определяется увеличение количества плазматических клеток и макрофагов, фагоцитирующих жиры. Содержание недифференцированных клеток – в пределах нормы. Снижена концентрация клеток миелоцитарного ростка, увеличен показатель лимфоцитов.
Лечение анемии Фанкони
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, зависят от тяжести заболевания, возраста пациента, наличия и выраженности врожденных аномалий. Дополнительно проводится лечение инфекций и онкопатологий, осуществляются реабилитационные мероприятия. Для устранения анемии используются следующие методы:
- ТКМ. Трансплантация костного мозга является наиболее эффективной в долгосрочной перспективе, но имеет противопоказания, нередко сопровождается развитием осложнений. Оптимальный возраст для проведения операции – до десяти лет. Донорами могут выступать здоровые сестра и братья, подходящие по критериям совместимости. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации сохраняется высокая вероятность острого или хронического иммунного конфликта между клетками донора и реципиента.
- Медикаментозная стимуляция кроветворения. При невозможности проведения трансплантации пациентам показано консервативное лечение, временно улучшающее их состояние. Выработка кровяных клеток стимулируется андрогенами (мужскими половыми гормонами) и гематопоэтическими факторами роста – эритропоэтином, фактором стволовых клеток, интерлейкинами-1-12. Параллельно применяются иммунодепрессанты. Медикаментозная терапия способна на протяжении многих лет поддерживать высокое качество жизни больных, но ее эффективность постепенно снижается.
- Переливание компонентов крови. При выраженных побочных эффектах или противопоказаниях к этиотропной терапии (трансплантации, стимуляции кроветворения) назначаются процедуры гемотрансфузии. Переливаются отмытые эритроциты – донорские красные кровяные тельца, освобожденные от поверхностных белков. При кровотечениях и снижении уровня тромбоцитов пациентам вводится тромбоцитарная масса.
Прогноз и профилактика
Продолжительность жизни больных определяется степенью нарушения функции костного мозга. Иногда пациенты доживают до 40 лет без лечения, но нередко умирают в детстве от тяжелой анемии или онкологических заболеваний. Прогноз наиболее благоприятен при своевременном проведении аллогенной трансплантации костного мозга, после которой есть шанс полного восстановления нормального кроветворения и увеличения срока жизни. Поскольку заболевание генетическое, предотвратить его развитие невозможно. Профилактика сводится к медико-генетической консультации супружеских пар из групп риска, планирующих беременность, а также к проведению пренатальной диагностики патологии, в ходе которой из пуповинной вены плода производится забор крови и выполняется ДЭБ-тест. При его положительном результате рассматривается вопрос о прерывании беременности.