Анемия с дефицитом пируваткиназы
К настоящему
времени накопилось достаточное число
наблюдений, позволяющих должным образом
оценить этиологическую роль генетической
недостаточности пируваткиназы в развитии
семейной, наследственной гемолитической
анемии «несфероцитарного» типа.
Описания современных
авторов, в частности Bowman
с соавторами, наблюдавших
21 семейный
случай пируваткиназа-дефицитной анемии
в одном изоляте, дают основание
рассматривать эту анемию как проявление
ферментной аномалии эритроцитов,
при которой нарушается внутриэритроцитарный
гликолиз на последних этапах образования
3-фосфоглицериновой кислоты из
1,3-дифосфоглицериновой кислоты и
образование пировиноградной кислоты
из фосфоэнолпировиноградной кислоты,
катализируемых пируваткиназой. Резкое
снижение активности пируваткиназы в
эритроцитах больных «несфероцитарной»
гемолитической анемии
II типа (по
Selvin
и
Dacie) приводит
к нарушению образования аденозин-трифосфорной
кислоты (АТФ); непосредственный же
механизм гемолиза, как полагают
современные авторы, обусловлен нарушением
электролитного баланса K/Na
(в котором АТФ играет основную роль) с
задержкой натрия в эритроцитах, что
приводит к снижению их осмотической
стойкости и гемолизу (корригируемому
АТФ).
Дефицит пируваткиназы
обнаруживается и у родителей больных,
но в меньшей степени, не приводящей
к явлениям гемолиза. Последний проявляется
только у гомозигот по данному признаку.
Аутогемолиз в
связи с нарушенным синтезом АТФ в
эритроцитах описан в единичных случаях
и при других ферментных недостаточностях:
дефиците триозофосфатизомеразы
(Schneider
с соавторами,
1965),
наследуемом по аутосомно-рецессивному
типу и проявляющемуся у гомозигот;
дефиците 2,3-дифосфоглицеромутазы,
наследуемом по аутосомно-доминантному
типу (Bowdler,
Prankerd,
1964).
Описаны также
больные «несфероцитарной» гемолитической
анемией, связанной с дефицитом
аденозинтрифосфатазы (Harvald
с соавторами,
1964),
дегидрогеназы 6-фосфоглюконовой кислоты
(Л. И. Идельсон и Г. В. Ермильченко,
1967).
Диагноз
энзимопенической («несфероцитарной»)
гемолитической анемии ставится на
основании клинико-гематологической
картины болезни и данных специальных
лабораторных проб, выявляющих
недостаточность ферментных систем
эритроцитов. Опорными пунктами в
дифференциальной диагностике с врожденной
гемолитической болезнью Минковского—Шоффара
служит макропланоцитоз при нормальной
или незначительно сниженной
осмотической резистентности эритроцитов.
Неизменно
отрицательная проба Кумбса и признаки
врожденного страдания позволяют легко
отличить данное заболевание от
приобретенной аутоиммунной
гемолитической анемии.
Прогноз.
Предсказание серьезно, особенно в период
гемолитических кризов.
Лечение.
Патогенетической терапии не существует.
В период гемолитических кризов
рекомендуется комплексная терапия
антианемическими препаратами (железо,
витамин B12)
и переливаниями эритроцитной массы.
Спленэктомия в большинстве случаев при
врожденной «несфероцитарной»
гемолитической анемии оказывалась
безрезультатной. Лишь в некоторых
случаях был получен частичный эффект.
По данным Ю. И. Лорие (1961),
обобщившего опыт ЦОЛИПК (всего 9
случаев) а также нашей клиники
(3 случая),
спленэктомия дает относительно
благоприятный результат в более
легких случаях заболевания (анемия
I типа по
Selvin
и Dacie),
характеризующихся менее выраженным
макроцитозом (средний диаметр эритроцитов
7,44—7,8
мкм) и нерезким снижением осмотической
стойкости эритроцитов после суточной
инкубации. В более тяжелых случаях
анемии
(II тип по
Selvin
и
Dacie),
характеризующихся более выраженным
макропланоцитозом (средний диаметр
эритроцитов
8,5— 8,88 мкм)
и резким снижением осмотической стойкости
эритроцитов после суточной инкубации,
спленэктомия не эффективна и даже
опасна. Аналогичные результаты получены
Grouchy
с сотрудниками
(1960),
На основании
сказанного выше вопрос о показаниях к
спленэктомии следует решать в соответствии
с клиническими данными (анемия, частые
гемолитические кризы, спленомегалия
со вторичным «гиперспленизмом»
—
лейкотромбоцитопенией) и руководствуясь
«типажем» эритроцитов (определяемым
по данным эритроцитометрии и осмотической
стойкости эритроцитов после суточной
инкубации без глюкозы и с добавлением
глюкозы). Решающее значение в прогнозе
операции, ее возможной эффективности
в данных случаях приобретают методы
радиоизотопной индикации при помощи
Cr51
и изучения приживаемости донорских
эритроцитов. В случае обнаружения
преобладающей секвестрации эритроцитов
в селезенке и сокращения сроков циркуляции
донорских эритроцитов (что может
указывать на существование
внеэритроцитарных факторов гемолиза)
можно рассчитывать на успех спленэктомии.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
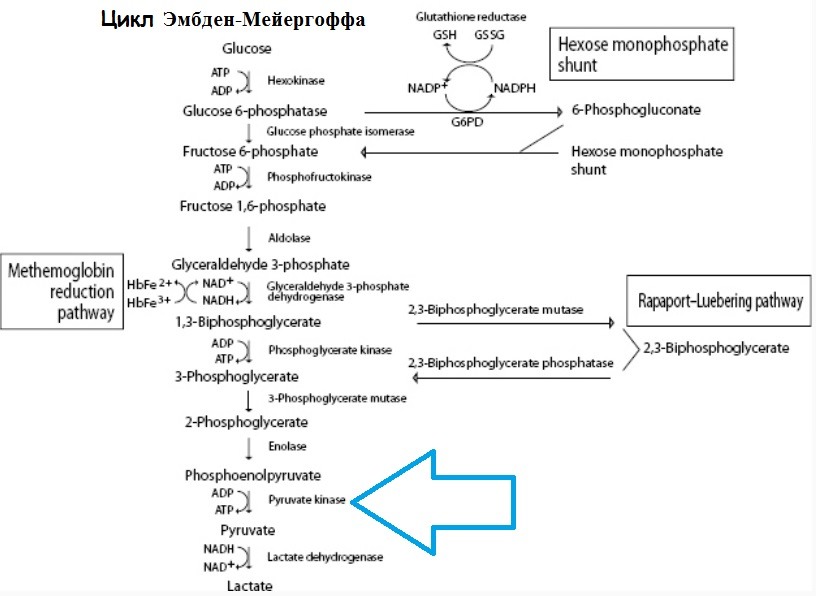
Различные дефекты ферментов эритроцитов вызывают гемолитические анемии, которые характеризуются отсутствием сфероцитов и некоторыми особенностями в мазке крови. Описаны случаи дефицита большей части ферментов как анаэробного пути Эмбдена-Мейергофа, так и окислительного гексозомонофосфатного (пентозного) шунта. Наиболее частым дефектом гликолитического фермента, который является причиной гемолитической анемии, является дефицит пируваткиназы, хотя само по себе это довольно редкое нарушение (известно всего 300-400 случаев).
Врожденная гемолитическая анемия наблюдается у лиц, гомозиготных по гену, который наследуется по аутосомно-рецессивному типу и вызывает значительное снижение активности пируваткиназы эритроцитов или продукцию аномального фермента с пониженной активностью. В эритроцитах нарушается выработка АТФ, снижается уровень АТФ, пирувата и образуется окисленная форма НАД+. Повышается концентрация 2,3-ДФГ, что, с одной стороны, способствует отдаче кислорода гемоглобином, с другой — ингибирует гексокиназу и ферменты гексозомонофосфатного шунта. Кроме того, происходит непонятное уменьшение совокупного количества адениннуклеотидов (АТФ, АДФ, АМФ) и пиридиннуклеотидов (НАД+ + НАДФ), что еще более нарушает гликолиз. При уменьшении генерации АТФ в эритроцитах нарушается нормальный баланс калия и воды; клетки обезвоживаются, становятся жесткими, и их продолжительность жизни значительно сокращается.
У млекопитающих существует два гена, кодирующих пируваткиназу, но только один ген (PKLR) экспрессирован в эритроцитах. Человеческий ген PKLR картирован на хромосоме lq21; известно 133 мутации в этом структурном гене, который кодирует белок, состоящий из 574 аминокислот, и образует функциональный тетрамер. Большинство пациентов являются компаундами (гетерозиготами по двум мутантным аллелям одного локуса) по двум разным дефектным генам пи-руваткиназы. Вариабельность клинического проявления в большой степени зависит от множества вероятных комбинаций.
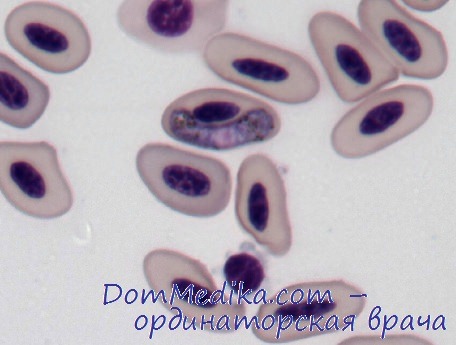
Клинические проявления дефицита пируваткиназы варьируются от тяжелой неонатальной гемолитической анемии до легкого компенсированного гемолиза, впервые проявляющегося во взрослом возрасте. В неонатальном периоде могут наблюдаться резко выраженные желтуха и анемия, известны случаи ядерной желтухи. У детей более старшего возраста и у взрослых гемолиз имеет разную степень с уровнем гемоглобина в пределах 8-12 г/дл, что ассоциируется с бледностью, желтушной окраской и спленомегалией. Этим пациентам трансфузия обычно не требуется. Тяжелая форма заболевания часто встречается среди аманитов, проживающих на Среднем Западе США.
Полихроматофилия и легкий макроцитоз обусловлены повышенным количеством ретикулоцитов. Сфероциты встречаются редко, но обычно обнаруживается несколько пикноцитов со спикулами. Осмотическая стойкость неинкубированных клеток нормальная. Аутогемолиз умеренно или значительно повышен, но добавление глюкозы не всегда корригирует эту аномалию, в отличие от наследственного сфероцитоза.
Диагноз дефицита пируваткиназы основан на значительном снижении активности пируваткиназы или увеличении диссоциации константы Михаэлиса—Ментена (Km) для субстрата этого фермента в эритроцитах (фосфоэнолпирувата). Активность других ферментов эритроцита нормальная или повышена в связи с ретикулоцитозом. Аномалий гемоглобина не отмечается. Поскольку пируваткиназа обладает нор мальной активностью в лейкоцитах, они должны быть полностью исключены из гемолизатов, используемых для оценки ее активности. У гетерозиготных носителей обычно наблюдается умеренно сниженный уровень активности пируваткиназы.
Лечение дефицита пируваткиназы
При гипербилирубинемии новорожденным показаны обменные трансфузии. При тяжелой анемии или при апластических кризах необходимы трансфузии эритроцитной массы. При постоянно тяжелой анемии или при необходимости частых трансфузий показана спленэктомия, которая выполняется в возрасте после 5-6 лет. Эта операция не излечивает заболевание, но после нее значительно повышается уровень гемоглобина и до удивительно высоких цифр повышается процентное содержание ретикулоцитов (30-60 %).
После спленэктомии может произойти гибель пациента от пневмококкового сепсиса; поэтому перед операцией необходимо провести иммунизацию вакцинами от инкапсулированных организмов, а после операции — пенициллинопрофилактику.
— Читать «Анемии при ферментопатиях у детей. Недостаточность ферментов гексозомонофосфатного пути»
Оглавление темы «Гемоглобинопатии у детей»:
- Аномальные гемоглобины у детей. Диагностика
- Наследственная метгемоглобинемия. Диагностика
- Синдромы персистирования фетального гемоглобина. Талассемия
- Гомозиготная b-талассемия — анемия Кули. Диагностика
- Лечение b-талассемии — анемии Кули
- B-талассемические синдромы. Промежуточная талассемия
- Альфа-талассемия у детей. Диагностика и лечение
- Гемохромотозы у детей. Избыточное накопление железа
- Дефицит пируваткиназы у детей. Диагностика и лечение
- Анемии при ферментопатиях у детей. Недостаточность ферментов гексозомонофосфатного пути
Дефицит эритрoцитарной пируваткиназы (PK deficiency) – состояние, приводящее к гемолитической анемии, взаимосвязанное с нарушением метаболизма в эритроцитах, изменением формы эритроцитов и нарушением их функционирования.
Первое описание заболевания датируется началом 90-х годов у кота абиссинской породы. А в 2000 году доктор Урз Гигер (Pen University) уже разработал генетический тест.
Дефицит эритроцитарной пируваткиназы передается по аутосомно-рецессивному типу наследования. То есть животное может являться носителем дефектного гена (быть гетерозиготным по данному гену) без проявления клинических симптомов заболевания. Скрещивание при разведении двух носителей будет давать 25% котят с дефицитом.
Уже к 2012 году были проведены многочисленные исследования, которые выявили дефектный ген во многих породах кошек. Наиболее часто встречается у представителей абиссинской породы, египетской мау и бенгальской. Также дефицит пируваткиназы был зарегистрирован у таких пород кошек, как: домашняя короткошёрстная и полудлиношерстная, ла перм, саванна, сибирская кошка, сингапурская, сомалийская.
По последним данным проведенных исследований в Америке доказано, что дефицит эритроцитарной пируваткиназы выявлен у большого количества других пород, в частности, мейн-кун и норвежская лесная.
Пируваткиназа — этот фермент, который катализирует превращение фосфоэнолпирувата в пируват и таким образом участвует в гликолитической реакции образования АТФ. При его дефиците развивается нестабильность эритроцитов, что в конечном итоге приводит к анемии. Данная патология в основном протекает неостро или же развивается постепенно, поэтому больное животное имеет возможность приспособиться к анемии и не демонстрировать характерные симптомы. По причине нечеткой клинической картины и вероятности проявления в любом возрасте без проведения генетических тестов это заболевание можно неправильно диагностировать.
Клинические признаки могут очень сильно варьироваться: от снижения аппетита с потерей веса до состояния серьезной летаргии. У некоторых животных может наблюдаться увеличение объема живота по причине увеличения размеров селезенки (спленoмегалия), бледность слизистых оболочек, повышение частоты сердечных сокращений (тахикардия). В редких случаях развивается желтушность, атрофия мышц. Данную патологию тяжело выявить по причине непостоянного проявления клинических симптомов. Организм больного животного чаще успевает возмещать некоторые проявления анемии, и, как результат, сложные случаи выявляются у возрастных животных. Диагностируются случаи проявления острого течения заболевания, в результате которого развивается тяжелая анемия.
При посещении ветеринарной клиники Вам нужно будет подробно рассказать врачу о здоровье своего питомца с начала проявления симптомов и про их характер. Затем ветеринарный врач произведет полное физиологическое обследование животного, а также забор анализов: полный биохимический и общий клинический анализы крови, анализ мочи (его желательно принести заблаговременно с собой).
Анализ крови может выявить увеличение числа тромбоцитов, а также белых кровяных клеток (лейкоцитоз), анемии с аномально большими, бледно-красными кровяными клетками, аномально сформированных эритроцитов, называемых пoйкилoцитами, и вариации цвета (пoлихромазия). Биохимические анализ крови чаще всего может показать избыток железа в крови (гиперферремия), незначительное рост уровня билирубина и незначительное повышение ферментов печени. По анализу мочи можно обнаружить повышение уровня билирубина.
Существует анализ ДНК на дефицит ПК, чтобы помочь владельцам и селекционерам идентифицировать кошек-носителей данного гена. В тесте используется материал, который несложно самостоятельно собрать у животных при помощи щёчных тампонов, во избежание инвазивного забора крови. Заводчики имеют возможность использовать данное тестирование животных во избежание скрещивания носителей между собой и проведения более тщательной селекционной работы.
Лечение.
Чаще всего ветеринарными специалистами применяется поддерживающая терапия.
При клиническом проявлении дефицита пируваткиназы острой анемией может быть рекомендовано проведение гематрансфузии. Существуют данные, что удаление селезенки (спленэктомия) может способствовать замедлению процесса разрушения эритроцитов.
Трансплантация костного мозга — единственное доступное лечение для кошек с дефицитом ПK. Однако это лечение дорого, потенциально опасно для жизни и не применяется в нашей стране.
Животные без лечения или с отсутствием ответа на лечение, как правило, умирают в возрасте до четырех лет в результате патологии костного мозга или печеночной недостаточности. У большинства из этих пациентов развивается тяжелая анемия и скопление жидкости в брюшной полости (асцит) во время терминальной стадии болезни.
Профилактикой является тестирование производителей и не допуск в разведение пораженных животных.
Если вы планируете покупать кошку из группы пород риска, всегда спрашивайте заводчика, были ли проведены тесты у производителей на дефицит ПК, и попросите посмотреть результаты.
При диагностировании у животного гомозиготного гена ПК-дефицита, рекомендуется обратиться к ветеринарному врачу для получения информации о прогрессировании заболевания и снижении тяжести течения болезни.
Недостаток пируваткиназы в эритроцитах — история изучения, причины, патофизиологияНедостаток пируваткиназы в эритроцитах заболевание, передающееся по наслеству. Оно характеризуется сокращением или исчезновением этого фермента из эритроцитов, в результате чего отмечается снижение способности сахарного обмена и концентрации АТФ, равно как и развитие хронической гемолитической анемии различной тяжести. В 1950 году были организованы исследования так называемых «несфероцитных врожденных гемолитических анемий». К тому же периоду был введен в практику тест аутогемолиза, заключающийся в 48-часовой инкубации, при 37°С дефибринированных в стерильных условиях эритроцитов в присутствии глюкозы или без нее и оценке самопроизвольного гемолиза. Применение этого теста в исследовании несфероцитной врожденной гемолитической анемии привело к выделению следующих двух групп: В отдельных случаях в этой второй категории отмечены частичная неспособность эритроцитов метаболизировать глюкозу до стадии лактата, также значительное накопление промежуточных метаболитов, в основном 2,3-дифосфоглицерата. В 1961 г. было установлено, что причину этого явления составляет отсутствие, в эритроцитах данных лиц, пируваткиназы, катализующей преобразование фосфоэнолпирувата в щавелевоуксусную кислоту. Со временем были описаны многие случаи врожденной гемолитической анемии за счет недостатка этого фермента, который хотя далеко не так часто встречается как дефицит Г-6-ФД, тем не менее находится на втором месте по частоте всех эритроэнзимопатий. Цикл Эмбден-Мейергоффа эритроцитов — последствия недостатка пируваткиназы Причины (этиология) недостатка пируваткиназы в эритроцитахПри дефиците пируваткиназы в эритроцитах в основе клинических нарушений находится генетическое поражение, которое проявляется либо сокращенным синтезом нормальных молекул, либо синтезом молекул неполноценной эффективности. Дефицит передается по наследству детям обоего пола, как рецессивная аутосомальная черта. Исследование пируваткиназы в иных клетках или тканях (лейкоцитах, мышцах, печени) выявило присутствие в них этого фермента, в нормальной концентрации, у страдающих недостатком пируваткиназы в эритроцитах, а это наводит на мысль о наличии ряда специализированных генов, каждый из них управляя синтезом определенного вида пируваткиназы. У гомозиготных больных наблюдается хроническая гемолитическая анемия и спленомегалия, в то время как у гетерозиготов клиническое и гематологическое состояние в норме. Большей частью нарушение носит количественный характер, однако описан ряд изоэнзим с качественными изменениями активности, что подтверждает наличие сдвигов в структурных генах. Патофизиология недостатка пируваткиназы в эритроцитахЖизнь эритроцита зависит от эффективности метаболизации глюкозы до ступени лактата, что составляет источник энергии, необходимой для проведения нормальной активности. Недостаток пируваткиназы препятствует осуществлению важной каталитической фазы, вырабатывающей энергию. Некоторое время молодые эритроциты работают с запасами ферментов, которыми располагают при поступлении в кровообращение но с возрастом разрушение ферментов, которые не могут быть восстановлены из-за отсутствия ядерного аппарата, равно как и блокирование метаболизма на уровне фосфоэнолпирувата определяют сокращение продолжительности жизни эритроидных элементов. Процесс блокирования может оказаться результатом не только большого сокращения численности молекул фермента, но также кинети.ческих аномалий. Так, некоторые изоэнзимы, пируваткиназы проявляют малое сродство к фосфоэнолпирувату, а это еще больше понижает эффективность молекул (и без того в уменьшенном, по сравнению с нормой числа) в процессе разрушения этого субстрата. В настоящее время еще не вскрыты полностью процессы, обусловливающие гемолиз эритроцитов с недостатком пируваткиназы, вместе с тем наблюдения многих авторов свидетельствуют об отсутствии постоянного и удовлетворительного взаимоотношения интенсивности дефицита и клинических проявлений. — Также рекомендуем «Течение, осложнения и лечение недостатка пируваткиназы в эритроцитах» Оглавление темы «Анемии»:
|