Анемия фанкони у детей клинические рекомендации
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Патогенез
- Симптомы
- Возможные осложнения
- Диагностика
- Лечение
- Список литературы
Другие названия и синонимы
Врожденная панмиелопатия Фанкони, Наследственная панмиелопатия.
Названия
Название: Анемия Фанкони.
Анемия Фанкони
Синонимы диагноза
Врожденная панмиелопатия Фанкони, Наследственная панмиелопатия.
Описание
Это генетическое заболевание, передающееся аутосомно-рецессивным путем, которое характеризуется нарушением кроветворения, образованием злокачественных опухолей, пороками развития и ломкостью хромосом. Это проявляется частым кровотечением, синяками, вялостью, бледностью и склонностью к инфекции. Диагностика проводится лабораторными методами, рекомендуются цитогенетические, генетические молекулярные и клинические анализы крови, миелограмма. Основными методами лечения являются трансплантация костного мозга, медицинское обеспечение кроветворения, переливание крови.
Анемия Фанкони
Дополнительные факты
Синонимами анемии Фанкони являются врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Болезнь получила свое название от швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов трех братьев. Анемия Фанкони является редким генетическим заболеванием, наследуемым по аутосомно-рецессивному принципу. Эпидемиологические показатели невысоки: на 1 больного ребенка приходится 350 тысяч младенцев. Распространенность одинакова между представителями мужского и женского пола; оно выше в общинах с разрешенными близкими родственниками, например, среди некоторых южноафриканских народов.
Причины
Болезнь наследственная; это развивается, когда дефектный ген передается от родителей ребенку. Было идентифицировано 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена расположен в Х-хромосоме (связан с полом). Все эти гены отвечают за выработку определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что отец и мать должны нести патологическую генетическую информацию. Кроме того, они вообще здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, которые получают один и тот же измененный ген от каждого родителя. В очень редких случаях анемия вызвана передачей дефектной Х-хромосомы. Женщины могут нести мутации, болезнь проявляется только у мальчиков. Риск развития патологии у сына, если у матери мутированный ген, составляет 50%.
Патогенез
Обычно в клетках организма существуют специальные ферментативные системы, которые корректируют разрывы молекул ДНК, поврежденных при биосинтезе или воздействии химических, физических реагентов. При анемии Фанкони в кластере белков, ответственных за репарацию ДНК, обнаружен генетический дефект, что приводит к повышенной ломкости хромосом. В результате у пациентов развивается дисфункция костного мозга — рак и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом — злокачественной опухолью костного мозга, которая вызывает накопление измененных лейкоцитов, которые ингибируют рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии рост и созревание всех трех типов клеток крови быстро ингибируются дисплазией костного мозга.
Симптомы
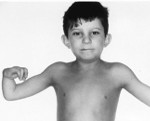
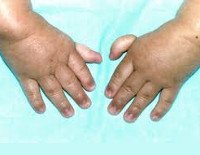
Более половины пациентов имеют врожденные аномалии внутренних органов и скелета. Деформации костей проявляются специфическим внешним видом: пациенты имеют низкорослый вид, уменьшенный размер головы, отсутствие или заметное укорочение большого пальца на руках, недоразвитие радиуса, врожденный вывих бедра и / или наличие шейного ребра, косолапость недоразвитый подбородок («птичье лицо»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, дрожанием глаз, глухотой, умственной отсталостью. Пациенты часто имеют незрелые половые органы, им не хватает одного или обоих яичек. Общие нарушения в структуре органов включают дефекты мочевыделительной системы: удвоение мочеточников или таза, подковообразные почки, почечные кисты, смещение наружного отверстия мочеиспускательного канала (гипоспадия). Врожденные пороки сердца включают атрезию трикуспидального клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Основные симптомы связаны с постепенным увеличением нарушений костного мозга. Чаще всего они дебютируют в детстве (от 5 до 10 лет). Из-за уменьшения количества тромбоцитов усиливается кровотечение: кровь не сгущается в течение длительного времени, носовые кровотечения возникают легко, выделения во время менструации обильные, много «беспричинных» синяков на теле. Снижение количества эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружением, обмороком, бледностью кожи, сердцебиением и одышкой. Дефицит лейкоцитов способствует ухудшению устойчивости к инфекциям. Впоследствии образуются лейкемия, миелодиспластический синдром и рак.
Вялость. Одышка.
Возможные осложнения
Частые инфекционные заболевания считаются наиболее распространенным осложнением. У пациентов развиваются ОРВИ, тонзиллит, ринит, бронхит, грипп, брюшной тиф и герпес. Рецидивирующий характер заболевания и его тяжелое течение приводят к разрушению органов, сопровождающемуся риском сепсиса. Еще одним осложнением наследственной анемии являются злокачественные новообразования — лейкемия, эпителиальные опухоли шеи и головы, половых органов. Рак у этих пациентов трудно поддается лечению из-за повышенной хрупкости и уменьшения восстановления ДНК. Это явление ограничивает применение лучевой терапии цитотоксическими препаратами. Нарушение свертываемости крови становится причиной большой потери крови.
Диагностика
Обследование пациентов проводится онкологами, гематологами, педиатрами и генетиками. Диагноз начинается с анализа анамнестических данных и жалоб. Врач выясняет, страдают ли эти родственники от этого наследственного заболевания, указывает время, в которое появляются первые признаки заболевания, ранние визиты к врачу. Во время обследования оценивается общее состояние пациента и выявляется наличие нарушений развития, гиперпигментированных пятен, кровотечений и кровоподтеков. В большинстве случаев нетрудно определить типичные деформации костей и недоразвитие глаз. Чтобы подтвердить диагноз и отличить анемию Фанкони от приобретенной анапластической анемии, проводится ряд лабораторных исследований:
• Клинический анализ крови. Изменения клеточного состава крови характерны. Тромбоцитопения и лейкопения диагностируются на ранних стадиях кроветворения, а панцитопения (резкое уменьшение объема эритроцитов, лейкоцитов и тромбоцитов) на поздних стадиях. Небольшой гемолиз возможен без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ возросло до 60-80 мм /.
• Цитогенетическое исследование клеток. Тест проводится с диэпоксибутаном, митомицином С, с указанием частоты и спектра хромосомных аберраций. В пользу генетической анемии параметры теста DEB считаются более 45%, пограничный уровень составляет 11-45% (процент клеток с хромосомными нарушениями).
• Молекулярно-генетический анализ клеток. Изучены гены, в которых мутации могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% в аллеле FANCC и в 10% в генах FANCG. Уровень мутаций в других парах составляет 0,2-3%. Согласно исследованию, установлено увеличение количества фагоцитарных жиров в плазматических клетках и макрофагах. Содержание недифференцированных клеток находится в пределах нормы. Концентрация зародышевых клеток миелоцитов была снижена, а количество лимфоцитов увеличено.
Лечение
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, в зависимости от тяжести заболевания, возраста пациента, наличия и тяжести врожденных дефектов. Кроме того, лечатся инфекции и онкопатологии и принимаются меры по реабилитации. Для устранения анемии используются следующие методы:
• BMT. Трансплантация костного мозга наиболее эффективна в отдаленном периоде, но имеет противопоказания, часто сопровождается развитием осложнений. Оптимальный возраст для операции — до десяти лет. Здоровые сестры и братья, которые соответствуют критериям отбора, могут быть донорами. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации вероятность острого или хронического иммунного конфликта между донорскими и реципиентными клетками остается высокой.
• Медикаментозная стимуляция кроветворения. Если трансплантация невозможна, пациентам показано консервативное лечение, которое временно улучшает их состояние. Продукцию клеток крови стимулируют андрогены (мужские половые гормоны) и гематопоэтические факторы роста — эритропоэтин, фактор стволовых клеток, интерлейкины-1-12. Параллельно используются иммунодепрессанты. Медикаментозная терапия на протяжении многих лет поддерживала высокое качество жизни пациентов, но ее эффективность постепенно снижалась.
• Переливание компонентов крови. При тяжелых побочных эффектах или противопоказаниях к этиотропной терапии (трансплантация, стимуляция кроветворения) назначаются процедуры переливания крови. Промывают эритроциты, донорские эритроциты освобождаются от поверхностных белков, переливаются. При кровотечении и снижении уровня тромбоцитов пациентам вводят массу тромбоцитов.
Список литературы
1. Генетическая диагностика анемии Фанкони. Обзор литературы/ Панферова А. В. Тимофеева Н. М. Ольшанская Ю. В. // Онкогематология. — 2016.
2. Анемии (клиника, диагностика, лечение). Методическое пособие/ Филатов Л. Б. — 2006.
3. Свободнорадикальный статус клеток крови и костного мозга у детей с острыми лейкозами и анемией Фанкони: Автореферат диссертации/ Чивилева И. Ю. — 1996. V.
4. Анемия Фанкони: современная диагностика и терапия: Автореферат дисертации/ Кравченко Е. Г. — 2003.

Анемия Фанкони – это генетическое заболевание, которое передается по аутосомно-рецессивному типу и характеризуется нарушением кроветворения, формированием злокачественных новообразований, пороками развития, ломкостью хромосом. Проявляется частыми кровотечениями, кровоподтеками на коже, вялостью, бледностью, склонностью к инфекциям. Диагностика проводится лабораторными методами, назначаются цитогенетическое, молекулярно-генетическое и клиническое исследования крови, миелограмма. Основные способы лечения – пересадка костного мозга, медикаментозное поддержание кроветворения, переливание крови.
Общие сведения
Синонимичные названия анемии Фанкони – врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Заболевание названо по фамилии швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов у трех братьев. Анемия Фанкони является редкой генетической болезнью, наследуется согласно аутосомно-рецессивному принципу. Эпидемиологические показатели низкие – 1 больной ребенок на 350 тысяч новорожденных. Распространенность одинакова среди представителей женского и мужского пола, выше в сообществах с разрешенными близкородственными браками, например, у некоторых южноафриканских народов.

Анемия Фанкони
Причины
Заболевание является наследственным, развивается при передаче дефектного гена от родителей к ребенку. Выявлено 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена находится в X-хромосоме (сцепленной с полом). Все эти гены отвечают за производство определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что и отец, и мать должны быть носителями патологической генетической информации. При этом сами они, как правило, здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, получивших от каждого из родителей один и тот же измененный ген. В крайне редких случаях анемия провоцируется передачей дефектной Х-сцепленной хромосомы. Женщины могут быть носительницами мутации, заболевание проявляется только у мальчиков. Риск развития патологии у сына при наличии у матери мутированного гена – 50%.
Патогенез
В норме в клетках организма существуют специальные ферментные системы, которые исправляют разрывы молекул ДНК, поврежденных в процессе биосинтеза или воздействия химических, физических реагентов. При анемии Фанкони обнаруживается генетический дефект в кластере белков, ответственных за репарацию ДНК, что приводит к повышенной ломкости хромосом. В итоге у пациентов развиваются нарушения функций костного мозга – неоплазии и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью миелоидного ростка крови, провоцирующей накопление измененных белых клеток, подавляющих рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии в результате дисплазии костного мозга резко угнетается рост и созревание всех трех видов клеток крови.
Симптомы анемии Фанкони
Более чем у половины пациентов наблюдаются врожденные аномалии развития внутренних органов и скелета. Костные деформации проявляются специфическим внешним видом: больные низкорослые, с уменьшенным размером головы, отсутствием или заметным укорочением большого пальца на руках, недоразвитием лучевой кости, врожденным вывихом бедра и/или наличием шейного ребра, косолапостью, недоразвитым подбородком («птичьим лицом»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, глазным дрожанием, глухотой, умственной отсталостью. Больные зачастую имеют незрелые половые органы, у них отсутствует одно или оба яичка. К распространенным аномалиям строения органов относятся пороки мочевыделительной системы: удвоение мочеточников или лоханки, подковообразные почки, почечные кисты, смещенное наружное отверстие уретры (гипоспадия). Врожденные пороки сердца включают атрезию трехстворчатого клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Ключевые симптомы связаны с постепенным нарастанием нарушений в работе костного мозга. Чаще они дебютируют в детском возрасте (в 5-10 лет). Из-за снижения количества тромбоцитов развивается повышенная кровоточивость: при ранениях кровь долго не сворачивается, легко возникают носовые кровотечения, выделения при менструациях обильны, на теле обнаруживается много «беспричинных» кровоподтеков. Уменьшение числа эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружениями, обмороками, бледностью кожи, учащенным сердцебиением и одышкой. Недостаток лейкоцитов способствует ухудшению сопротивляемости инфекциям. Впоследствии формируется лейкоз, миелодиспластический синдром, онкологические болезни.
Осложнения
Наиболее распространенным осложнением считаются частые инфекционные заболевания. У пациентов развивается ОРВИ, ангина, ринит, бронхит, грипп, тиф, герпес. Рецидивирующий характер болезней и их тяжелое течение приводят к деструкции органов, сопровождаются риском сепсиса. Другим осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли органов шеи и головы, половых органов. Рак у таких больных тяжело поддается лечению из-за повышенной ломкости и сниженной репарации ДНК. Это явление ограничивает применение лучевой терапии, цитотоксических препаратов. Нарушение свертываемости становится причиной больших кровопотерь.
Диагностика
Обследование больных проводят онкологи, гематологи, педиатры, врачи-генетики. Диагностика начинается с анализа анамнестических данных и жалоб. Врач выясняет, имеется ли данное наследственное заболевание у близких родственников, уточняет время появления первых признаков болезни, ранние обращения к врачам. При осмотре оценивает общее состояние пациента, выявляет наличие аномалий развития, гиперпигментированных пятен, кровотечений, кровоподтеков. В большинстве случаев не составляет труда обнаружить типичные деформации костей, недоразвитие глаз. Для подтверждения диагноза и различения анемии Фанкони с приобретенной анапластической анемией проводится ряд лабораторных исследований:
- Клинический анализ крови. Характерны изменения клеточного состава крови. На ранних этапах нарушения кроветворения диагностируется тромбоцитопения и лейкопения, на более поздних – панцитопения (резкое снижение объема эритроцитов, лейкоцитов и тромбоцитов). Возможен умеренный гемолиз без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ увеличено до 60-80 мм/ч.
- Цитогенетическое исследование клеток. Выполняется проба с диэпоксибутаном, митомицином C, указывающая на частоту и спектр хромосомных аберраций. В пользу генетической анемии рассматриваются показатели ДЭБ-теста более 45%, пограничный уровень – 11-45% (процент клеток с хромосомными разрывами).
- Молекулярно-генетический анализ клеток. Исследуются гены, мутации в которых могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% – в аллели FANCC, в 10% – в генах FANCG. Частота мутаций в других парах – 0,2-3%.
- Миелограмма. По данным исследования определяется увеличение количества плазматических клеток и макрофагов, фагоцитирующих жиры. Содержание недифференцированных клеток – в пределах нормы. Снижена концентрация клеток миелоцитарного ростка, увеличен показатель лимфоцитов.
Лечение анемии Фанкони
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, зависят от тяжести заболевания, возраста пациента, наличия и выраженности врожденных аномалий. Дополнительно проводится лечение инфекций и онкопатологий, осуществляются реабилитационные мероприятия. Для устранения анемии используются следующие методы:
- ТКМ. Трансплантация костного мозга является наиболее эффективной в долгосрочной перспективе, но имеет противопоказания, нередко сопровождается развитием осложнений. Оптимальный возраст для проведения операции – до десяти лет. Донорами могут выступать здоровые сестра и братья, подходящие по критериям совместимости. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации сохраняется высокая вероятность острого или хронического иммунного конфликта между клетками донора и реципиента.
- Медикаментозная стимуляция кроветворения. При невозможности проведения трансплантации пациентам показано консервативное лечение, временно улучшающее их состояние. Выработка кровяных клеток стимулируется андрогенами (мужскими половыми гормонами) и гематопоэтическими факторами роста – эритропоэтином, фактором стволовых клеток, интерлейкинами-1-12. Параллельно применяются иммунодепрессанты. Медикаментозная терапия способна на протяжении многих лет поддерживать высокое качество жизни больных, но ее эффективность постепенно снижается.
- Переливание компонентов крови. При выраженных побочных эффектах или противопоказаниях к этиотропной терапии (трансплантации, стимуляции кроветворения) назначаются процедуры гемотрансфузии. Переливаются отмытые эритроциты – донорские красные кровяные тельца, освобожденные от поверхностных белков. При кровотечениях и снижении уровня тромбоцитов пациентам вводится тромбоцитарная масса.
Прогноз и профилактика
Продолжительность жизни больных определяется степенью нарушения функции костного мозга. Иногда пациенты доживают до 40 лет без лечения, но нередко умирают в детстве от тяжелой анемии или онкологических заболеваний. Прогноз наиболее благоприятен при своевременном проведении аллогенной трансплантации костного мозга, после которой есть шанс полного восстановления нормального кроветворения и увеличения срока жизни. Поскольку заболевание генетическое, предотвратить его развитие невозможно. Профилактика сводится к медико-генетической консультации супружеских пар из групп риска, планирующих беременность, а также к проведению пренатальной диагностики патологии, в ходе которой из пуповинной вены плода производится забор крови и выполняется ДЭБ-тест. При его положительном результате рассматривается вопрос о прерывании беременности.

