Врожденная апластическая анемия фанкони

Анемия Фанкони – это генетическое заболевание, которое передается по аутосомно-рецессивному типу и характеризуется нарушением кроветворения, формированием злокачественных новообразований, пороками развития, ломкостью хромосом. Проявляется частыми кровотечениями, кровоподтеками на коже, вялостью, бледностью, склонностью к инфекциям. Диагностика проводится лабораторными методами, назначаются цитогенетическое, молекулярно-генетическое и клиническое исследования крови, миелограмма. Основные способы лечения – пересадка костного мозга, медикаментозное поддержание кроветворения, переливание крови.
Общие сведения
Синонимичные названия анемии Фанкони – врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Заболевание названо по фамилии швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов у трех братьев. Анемия Фанкони является редкой генетической болезнью, наследуется согласно аутосомно-рецессивному принципу. Эпидемиологические показатели низкие – 1 больной ребенок на 350 тысяч новорожденных. Распространенность одинакова среди представителей женского и мужского пола, выше в сообществах с разрешенными близкородственными браками, например, у некоторых южноафриканских народов.

Анемия Фанкони
Причины
Заболевание является наследственным, развивается при передаче дефектного гена от родителей к ребенку. Выявлено 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена находится в X-хромосоме (сцепленной с полом). Все эти гены отвечают за производство определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что и отец, и мать должны быть носителями патологической генетической информации. При этом сами они, как правило, здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, получивших от каждого из родителей один и тот же измененный ген. В крайне редких случаях анемия провоцируется передачей дефектной Х-сцепленной хромосомы. Женщины могут быть носительницами мутации, заболевание проявляется только у мальчиков. Риск развития патологии у сына при наличии у матери мутированного гена – 50%.
Патогенез
В норме в клетках организма существуют специальные ферментные системы, которые исправляют разрывы молекул ДНК, поврежденных в процессе биосинтеза или воздействия химических, физических реагентов. При анемии Фанкони обнаруживается генетический дефект в кластере белков, ответственных за репарацию ДНК, что приводит к повышенной ломкости хромосом. В итоге у пациентов развиваются нарушения функций костного мозга – неоплазии и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью миелоидного ростка крови, провоцирующей накопление измененных белых клеток, подавляющих рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии в результате дисплазии костного мозга резко угнетается рост и созревание всех трех видов клеток крови.
Симптомы анемии Фанкони
Более чем у половины пациентов наблюдаются врожденные аномалии развития внутренних органов и скелета. Костные деформации проявляются специфическим внешним видом: больные низкорослые, с уменьшенным размером головы, отсутствием или заметным укорочением большого пальца на руках, недоразвитием лучевой кости, врожденным вывихом бедра и/или наличием шейного ребра, косолапостью, недоразвитым подбородком («птичьим лицом»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, глазным дрожанием, глухотой, умственной отсталостью. Больные зачастую имеют незрелые половые органы, у них отсутствует одно или оба яичка. К распространенным аномалиям строения органов относятся пороки мочевыделительной системы: удвоение мочеточников или лоханки, подковообразные почки, почечные кисты, смещенное наружное отверстие уретры (гипоспадия). Врожденные пороки сердца включают атрезию трехстворчатого клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Ключевые симптомы связаны с постепенным нарастанием нарушений в работе костного мозга. Чаще они дебютируют в детском возрасте (в 5-10 лет). Из-за снижения количества тромбоцитов развивается повышенная кровоточивость: при ранениях кровь долго не сворачивается, легко возникают носовые кровотечения, выделения при менструациях обильны, на теле обнаруживается много «беспричинных» кровоподтеков. Уменьшение числа эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружениями, обмороками, бледностью кожи, учащенным сердцебиением и одышкой. Недостаток лейкоцитов способствует ухудшению сопротивляемости инфекциям. Впоследствии формируется лейкоз, миелодиспластический синдром, онкологические болезни.
Осложнения
Наиболее распространенным осложнением считаются частые инфекционные заболевания. У пациентов развивается ОРВИ, ангина, ринит, бронхит, грипп, тиф, герпес. Рецидивирующий характер болезней и их тяжелое течение приводят к деструкции органов, сопровождаются риском сепсиса. Другим осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли органов шеи и головы, половых органов. Рак у таких больных тяжело поддается лечению из-за повышенной ломкости и сниженной репарации ДНК. Это явление ограничивает применение лучевой терапии, цитотоксических препаратов. Нарушение свертываемости становится причиной больших кровопотерь.
Диагностика
Обследование больных проводят онкологи, гематологи, педиатры, врачи-генетики. Диагностика начинается с анализа анамнестических данных и жалоб. Врач выясняет, имеется ли данное наследственное заболевание у близких родственников, уточняет время появления первых признаков болезни, ранние обращения к врачам. При осмотре оценивает общее состояние пациента, выявляет наличие аномалий развития, гиперпигментированных пятен, кровотечений, кровоподтеков. В большинстве случаев не составляет труда обнаружить типичные деформации костей, недоразвитие глаз. Для подтверждения диагноза и различения анемии Фанкони с приобретенной анапластической анемией проводится ряд лабораторных исследований:
- Клинический анализ крови. Характерны изменения клеточного состава крови. На ранних этапах нарушения кроветворения диагностируется тромбоцитопения и лейкопения, на более поздних – панцитопения (резкое снижение объема эритроцитов, лейкоцитов и тромбоцитов). Возможен умеренный гемолиз без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ увеличено до 60-80 мм/ч.
- Цитогенетическое исследование клеток. Выполняется проба с диэпоксибутаном, митомицином C, указывающая на частоту и спектр хромосомных аберраций. В пользу генетической анемии рассматриваются показатели ДЭБ-теста более 45%, пограничный уровень – 11-45% (процент клеток с хромосомными разрывами).
- Молекулярно-генетический анализ клеток. Исследуются гены, мутации в которых могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% – в аллели FANCC, в 10% – в генах FANCG. Частота мутаций в других парах – 0,2-3%.
- Миелограмма. По данным исследования определяется увеличение количества плазматических клеток и макрофагов, фагоцитирующих жиры. Содержание недифференцированных клеток – в пределах нормы. Снижена концентрация клеток миелоцитарного ростка, увеличен показатель лимфоцитов.
Лечение анемии Фанкони
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, зависят от тяжести заболевания, возраста пациента, наличия и выраженности врожденных аномалий. Дополнительно проводится лечение инфекций и онкопатологий, осуществляются реабилитационные мероприятия. Для устранения анемии используются следующие методы:
- ТКМ. Трансплантация костного мозга является наиболее эффективной в долгосрочной перспективе, но имеет противопоказания, нередко сопровождается развитием осложнений. Оптимальный возраст для проведения операции – до десяти лет. Донорами могут выступать здоровые сестра и братья, подходящие по критериям совместимости. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации сохраняется высокая вероятность острого или хронического иммунного конфликта между клетками донора и реципиента.
- Медикаментозная стимуляция кроветворения. При невозможности проведения трансплантации пациентам показано консервативное лечение, временно улучшающее их состояние. Выработка кровяных клеток стимулируется андрогенами (мужскими половыми гормонами) и гематопоэтическими факторами роста – эритропоэтином, фактором стволовых клеток, интерлейкинами-1-12. Параллельно применяются иммунодепрессанты. Медикаментозная терапия способна на протяжении многих лет поддерживать высокое качество жизни больных, но ее эффективность постепенно снижается.
- Переливание компонентов крови. При выраженных побочных эффектах или противопоказаниях к этиотропной терапии (трансплантации, стимуляции кроветворения) назначаются процедуры гемотрансфузии. Переливаются отмытые эритроциты – донорские красные кровяные тельца, освобожденные от поверхностных белков. При кровотечениях и снижении уровня тромбоцитов пациентам вводится тромбоцитарная масса.
Прогноз и профилактика
Продолжительность жизни больных определяется степенью нарушения функции костного мозга. Иногда пациенты доживают до 40 лет без лечения, но нередко умирают в детстве от тяжелой анемии или онкологических заболеваний. Прогноз наиболее благоприятен при своевременном проведении аллогенной трансплантации костного мозга, после которой есть шанс полного восстановления нормального кроветворения и увеличения срока жизни. Поскольку заболевание генетическое, предотвратить его развитие невозможно. Профилактика сводится к медико-генетической консультации супружеских пар из групп риска, планирующих беременность, а также к проведению пренатальной диагностики патологии, в ходе которой из пуповинной вены плода производится забор крови и выполняется ДЭБ-тест. При его положительном результате рассматривается вопрос о прерывании беременности.
Источник
Апластическая анемия Фанкони – это редкое генетическое заболевание, вызванное мутацией некоторых генов, что можно обнаружить ещё в пренатальной стадии. Как это проявляется? Какие существуют методы лечения?
Описание и характеристики анемии Фанкони
Анемия Фанкони – это генетическое заболевание, характеризующееся панцитопенией, а именно – недостаточным производством со стороны спинного мозга клеток крови: эритроцитов, лейкоцитов и тромбоцитов.

Эти особенности повышают для больных анемией Фанкони риск развития серьезных осложнений, таких как опухоли и лейкоз.
Как передается анемия Фанкони
Болезнь передается генетически, по аутосомно-рецессивному пути, то есть человек заболеет, только если получит измененные гены от обоих родителей (заболевание возникает из-за мутации на одном гене, но он состоит из двух аллелей, одна наследуется от отца, другая от матери).
Заболевание более часто встречается в условиях тесного инбридинга и этнических сообществах. Типичным примером, в этом смысле, являются евреи ашкенази (потомки еврейских общин, которые в эпоху Средневековья жили в долине Рейна).
Обычно, болезнь проявляется при рождении в форме физических отклонений. Если симптомы отсутствуют при рождении, то появляются в возрасте от двух до пятнадцати лет.
Причины анемии Фанкони
Как я уже сказал, болезнь имеет генетический характер или вызвана изменения генов. Были обнаружены 13 генов на разных хромосомах, мутации которых ведут к аномалиям в процессе репарации ДНК.
Дефектные гены вызывают состояние аплазии костного мозга, точнее недостаточное производство клеток крови в костном мозге.
Симптомы и последствия анемии Фанкони
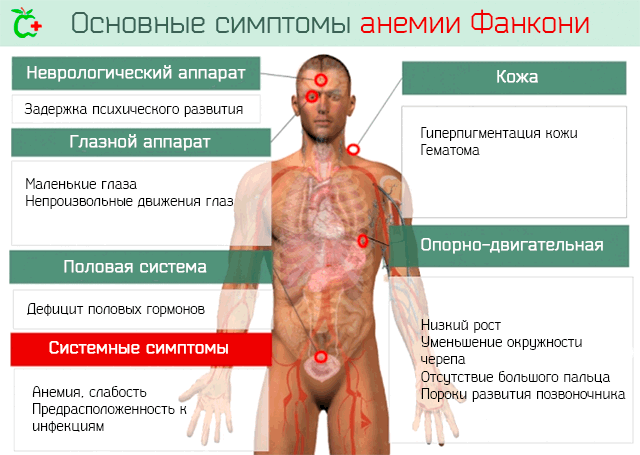
Симптомы, которыми проявляется анемия Фанкони:
- Многие пациенты имеют соматические нарушения: недостаточный вес при рождении, низкий рост, уменьшение окружности головы, маленькие глаза.
- Аномалии костей: отсутствие пальцев на руке или костей руки, пороки развития позвоночника, тазобедренного сустава или зубов.
- Аномалии органов: могут быть повреждены сердце, почки, пищевод, уши (с изменениями внутреннего уха, приводящими к глухоте).
- Недостаточная секреция половых гормонов.
- Гиперпигментация кожи с пятнами светло-коричневого цвета. Кожа становится темнее, чем обычно.
- Задержки психического развития.
- Ритмические и непроизвольные движения глаз. Расстройство, которое влияет на зрение.
Кроме того, по мере того как заболевание приводит к аплазии костного мозга с недостаточной продукцией клеток крови, это ведёт к появлению таких симптомов, как:
- Усталость, слабость и бледность.
- Увеличенная восприимчивость к инфекциям при снижении уровня лейкоцитов.
- Частое появление гематом и кровотечений, в случае уменьшения количество тромбоцитов.
Последствия
Обычно, заболевание даёт очень плохой прогноз и приводит к снижению продолжительности жизни тех, кто от него страдает. Предполагается, что средняя выживаемость составляет около 16-18 лет.
Те, кто страдает от анемии Фанкони, должны проходить постоянные проверки, так как из-за нестабильности хромосом с высокой долей вероятности могут развиваться:
- лейкоз
- некоторые формы раковых заболеваний
- кровоизлияния головного мозга, вызванные дефицитом тромбоцитов
Диагностика анемии Фанкони
Ранняя диагностика анемии Фанкони является очень важной и осуществляется через:
- полный анализ крови: подсчет эритроцитов, лейкоцитов, тромбоцитов, количества гемоглобина в крови
- изучение клеток (тест DEB). Позволяет выявить повреждения ДНА, что является признаком заболевания.
- В случае беременных женщин необходим амниоцентез для выявления возможных генетических дефектов у плода.
Терапия анемии Фанкони
Полное излечение заболевания возможно только одним путём – трансплантацией костного мозга.
Все переливания используются только для решения чрезвычайной ситуации.
Также некоторые лекарства, такие как эритропоэтин, так называемые факторы роста, помогают временно восстановить баланс в крови, так как увеличивают уровень эритроцитов и гемоглобина.
В последние годы, однако, открылась новая возможность, которая заключается в трансплантации стволовых клеток, которые могут делиться и давать начало новой линии клеток, способных дифференцироваться, давая происхождение всем клеткам крови. Стволовые клетки получают из пуповины подходящего донора.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 21 января 2019;
проверки требуют 2 правки.
Апласти́ческая анеми́я — заболевание кроветворной системы, характеризуется угнетением кроветворной функции костного мозга и проявляется недостаточным образованием эритроцитов, лейкоцитов и тромбоцитов (пангемоцитопенией) или только одних эритроцитов (парциальная гипопластическая анемия, синонимы: апластическая анемия, арегенераторная анемия, геморрагическая алейкия, миелопарез, миелофтиз, панмиелофтиз, прогрессирующая гипоцитемия). Для апластических анемий характерна выраженная панцитопения — анемия, лейкопения, тромбоцитопения и лимфопения[3].
История заболевания[править | править код]
Впервые это заболевание было описано Паулем Эрлихом в 1888 году у 21-летней женщины. Термин «апластическая анемия» был предложен Чауфордом в 1904 году. Апластическая анемия — одно из самых тяжёлых расстройств гемопоэза. Без лечения больные тяжёлыми формами апластической анемии погибают в течение нескольких месяцев. При своевременном адекватном лечении прогноз достаточно хороший. Длительный период времени апластическая (гипопластическая) анемия рассматривалась как синдром, объединяющий патологические состояния костного мозга протекающие с выраженной гипоплазией кроветворения. Современная медицина относит апластическую анемию к арегенераторному виду анемий (гипо-, апластические анемии)[4].
Этиология[править | править код]
Причинами апластической анемии могут быть:
- Химические вещества (мышьяк, ароматические углеводороды, в частности бензол, соли тяжёлых металлов).
- Ионизирующее излучение (см. Мария Склодовская-Кюри)
- Лекарственные препараты (НПВС, цитостатики, мерказолил, анальгин, левомицетин).
- Инфекционные агенты (вирусы, м/о).
- Аутоиммунные процессы (СКВ, синдром Шегрена).
Патогенез[править | править код]
Апластическая анемия может развиться при воздействии ряда миелотоксических факторов: ионизирующего излучения, химических веществ — бензола, солей золота, мышьяка; лекарственных средств — хлорамфеникола (левомицетина), фенилбутазона (бутадион), хлорпромазина (аминазин), мепробамата, дилантина, антиметаболитов (6-меркаптопурина, метотрексата), алкилирующих (циклофосфана, хлорбутина) и некоторых других средств. Миелотоксический эффект от воздействия одних факторов (ионизирующее излучение, антиметаболиты) возникает всегда при достаточно большой дозе, других — проявляется индивидуально. Причина индивидуальной чувствительности, в частности к некоторым лекарственным средствам не всегда ясна, но может быть связана с генетическими дефектами кроветворных клеток. Это относится, например, к хлорамфениколу и фенилбутазону, которые вызывают супрессию (в зависимости от дозы) эритропоэза с частотой соответственно 1:24000 и 1:40000 лиц, их принимающих.
Наследственный характер индивидуальной чувствительности эритропоэтических клеток к данным лекарственным веществам подтверждается развитием аплазии костного мозга у разных членов одной семьи и у однояйцевых близнецов. В других случаях вероятна связь индуцированного лекарственными веществами угнетения кроветворения с иммунными механизмами появлением антител к эритроцитарным предшественникам. Описаны случаи возникновения апластической анемии после острого вирусного гепатита (возможно, вследствие способности вируса гепатита изменять кариотип клеток, что было прослежено на культуре лейкоцитов), перенесенной инфекции вирусом Эпштейна — Барр, парвовирусом.
Существует и наследственная форма апластической анемии — анемия Фанкони.
Более чем у половины больных не удается выявить какие-либо причинные факторы — это так называемая идиопатическая апластическая анемия. Механизмы, лежащие в основе идиопатической формы анемии, неясны. Возможен аутоиммунный механизм, связанный с воздействием на клетки костного мозга аутоантител при участии иммунных лимфоцитов. Показано, что лимфоциты (Т-супрессоры) больных тормозят образование эритроцитных колоний костного мозга донора и могут нарушать дифференциацию и пролиферацию гематопоэтических предшественников.
Предполагают также, что основой апластической анемии может быть поражение (внутренний дефект) стволовой клетки, о чём свидетельствует восстановление кроветворения у больных после трансплантации им аллогенного костного мозга, содержащего нормальные стволовые клетки. Существуют экспериментальные данные, свидетельствующие о значении для развития апластического процесса и нарушений микроокружения — первичного дефекта стромальных клеток костного мозга. Однако суть этих клеточных дефектов остается неясной, так же как и их первичность. Возможно, что при разных формах апластической анемии патогенетические механизмы неодинаковы.
Клиника[править | править код]
- Анемический синдром (головокружение, снижение работоспособности, утомляемость, бледность кожных покровов и слизистых, сердцебиение, непереносимость длительных физических нагрузок и т. д.)
- Геморрагический синдром (кровоточивость, склонность к диапедезам, геморрагии)
- Инфекционные осложнения.
Диагностика[править | править код]
Картина периферической крови представлена трицитопенией. Снижение гемоглобина значительно и может достигать критического уровня 20 — 30 г/л. Цветовой показатель обычно равен единице, но в ряде случаев может быть гиперхромия и макроцитоз эритроцитов. Количество ретикулоцитов резко снижено. Характерна выраженная лейкопения (агранулоцитоз). Абсолютное содержание лимфоцитов не изменено или снижено. Количество тромбоцитов всегда снижено, в некоторых случаях не удается обнаружить их вообще. В большинстве случаев увеличивается СОЭ (до 40 — 60 мм/час).
Клиническая картина заболевания позволяет сформировать первичное представление о патологии системы крови. Отправной точкой диагностического поиска является клиническое исследование крови с подсчетом количества ретикулоцитов и тромбоцитов. Выявление би- или трицитопении при исследовании периферической крови служит основанием для выполнения морфологического исследования костного мозга.
Диагноз АА устанавливают на основании типичной гистологической картины костного мозга, получаемого методом трепанобиопсии гребня подвздошной кости. Для получения качественного (информативного) биоптата используются трепаны, выпускаемые промышленным способом (Sherwood medical).
При гистологическом исследовании костного мозга обнаруживается большое количество жировой ткани, содержание которой может достигать 90 %. Среди доминирующей жировой ткани встречаются стромальные и лимфоидные элементы. Гематогенные клетки представлены крайне скудно: в небольшом количестве встречаются эритроидные и гранулоцитарные предшественники. Мегакариоциты отсутствуют.
Лечение[править | править код]
Лечение апластической анемии представляет собой очень сложную задачу.
- Лечение с глюкокортикоидами эффективно, если болезнь обусловлена аутоиммунными механизмами, появлением антител против клеток крови.
- Лечение анаболическими препаратами стимулируют кроветворение.
- Лечение андрогенами обладает анаболическим эффектом и стимулируют эритропоэз.
- Лечение цитостатиками (иммунодепресантами) — назначается лишь при отсутствии эффекта от других методов лечение у больных с аутоиммунной формой, в том числе при парциальной красноклеточной аплазии.
- Спленэктомия
- Лечение антилимфоцитарным глобулином рекомендуется при отсутствии эффекта от спленэктомии и других методов лечения.
- Лечение циклоспорином. Циклоспорин А (сандиммун) обладает иммунодепрессантным эффектом, селективно ингибирует транскрипцию гена интерлейкина-2 в Т-лимфоцитах, подавляет продукцию Гамма интерферона и альфа фактора некроза опухоли.
- Трансплантация костного мозга.
Основным и единственным патогенетическим методом лечения апластической анемии, позволяющим рассчитывать на спасение жизни больного, является трансплантация костного мозга от совместимого донора.
При невозможности подобрать донора проводится паллиативная терапия. В качестве базисного препарата используется иммунодепрессант циклоспорин А. У больных нетяжёлой апластической анемией использование данного препарата позволяет рассчитывать в ряде случаев на успех. Кроме того использование циклоспорина А целесообразно и с тех позиций, что глюкокортикоиды, андрогены и антилимфоцитарный глобулин способны улучшить состояние гемопоэза у больных нетяжёлой апластической анемией, но, однако, при этом следует принимать во внимание повышенный риск развития в последующем клональных заболеваний костного мозга. Применение циклоспорина А сводит такой риск к минимуму. Следует также отметить, что у части больных нетяжёлой апластической анемией, преодолевших 6-месячный порог выживаемости, может наступить спонтанное улучшение даже если им не проводилось никакой иммуносупрессивной терапии. Эффект от иммуносупрессивной терапии у больных тяжёлой и крайне тяжелой апластической анемией сомнителен.
- Лечение колониестимулирующими факторами или миелоидными факторами роста — эти гликопротеиды, стимулирующие пролиферацию и дифференциацию клеток-предшественниц гемопоэза различных типов.
- Трансфузии эритроцитов; показания: выраженная анемия, гипоксия мозга, гемодинамические нарушения.
Все больные апластической анемией нуждаются в заместительной трансфузионной терапии эритроцитарной и/или тромбоцитарной массой. Объём трансфузионной терапии определяется показателями периферической крови и клиническими проявлениями заболевания. Кроме того, проводится антибактериальная и микостатическая терапия с целью профилактики или лечения инфекционных осложнений.
Прогноз[править | править код]
Ремиссию удаётся получить примерно у половины больных. Прогноз несколько лучше у детей, чем у взрослых. Наличие большого количества жира в костном мозге не говорит о необратимости процесса. Бывают случаи, когда и у таких больных наступает полная ремиссия и полная репарация костномозгового кроветворения. Прогноз лучше, когда увеличено содержание ретикулоцитов, когда в костном мозге имеется более полиморфная картина, когда имеется небольшое увеличение размеров селезёнки и хотя бы небольшой, но чёткий эффект от кортикостероидных гормонов. В этих случаях спленэктомия оказывает чаще хороший эффект вплоть до полного выздоровления. У части больных апластический синдром является началом острого лейкоза. Иногда признаки гемобластоза выявляются лишь через несколько лет от начала болезни.
Примечания[править | править код]
Источник
