В12 фолиеводефицитная анемия этиология патогенез
B12-дефицитная и фолиеводефицитная анемия — это анемии связанные с нарушением синтеза нуклеиновых кислот и заменой нормобластического типа кроветворения мегалобластическим из-за недостатка в организме витамина B12 и фолиевой кислоты.
Этиология.
1. Недостаток витамина в пище.
2. Неусвоение витамина B12 в желудке, что может быть связано с нарушением функции фундального отдела желудка, который вырабатывает гастромукопротеин (витамин B12 усваивается в комплексе с гастромукопртеином). Нарушение функции обкладочных клеток вызывается воздействием на них аутоантител (пернициозная анемия или Аддисона-Бирмера или злокачественное малокровие). Кроме того, подобное состояние может возникнуть после резекции желудка.
3. Неусвоение витамина B12 в кишечнике (при резекции тонкой кишки, опухоли, спру, дифиллоботриозе, алкоголизме).
4. Повышенное расходование витаминов при беременности.
5. Нарушение депонирования витаминов в печени при ее диффузном поражении.
Патогенез. Дефицит витамина B12 и фолиевой кислоты, участвующих в образовании тимина, входящего в состав ДНК, снижает скорость ее образования. Замедление репликации ДНК прежде всего заметно в тканях, где в норме деление клеток происходит наиболее часто — в клетках крови и эпителия желудочно-кишечного тракта. Нарушение клеточного деления приводит к формированию крупных клеток крови: мегалоцитов, мегалобластов, гигантских мегакариоцитов. Созревание мегалобластов до мегалоцитов сопровождается нарушением энуклеации (об этом свидетельствуют появление в мегалоцитах телец Жолли (остатки ядра) и колец Кебота (остатки ядерной облочки)). Наличие большого количества мегалобластов и мегалоцитов, насыщенных гемоглобином, обуславливает гиперхромию (ЦП>1.0).
Обычное физиологическое слущивание эпителия ЖКТ из-за нарушения клеточного деления не восстанавливается. Поэтому развиваются атрофически-воспалительные процессы в эпителии всего ЖКТ. При этом всасывание витаминов еще более нарушается.
В результате недостатка витамина B12 в организме накапливается метилмалоновая кислота, которая токсична для нервных клеток. Кроме того, при дефиците витамина B12 в нервных волокнах синтезируются жирные кислоты с измененной структурой, что отражается на образовании миелина и приводит к повреждению аксона. Развивается дегенерация задних и боковых столбов спинного мозга (фуникулярный миелоз), поражаются черепно-мозговые и периферические нервы.
Картина крови. B12-дефицитная и фолиеводефицитная анемия — это анемии мегалобластические, гиперхромные, макроцитарные. В мазке крови появляются мегалоциты — клетки патологической регенерации костного мозга и мегалобласты (крупные клетки с базофильной, полихроматофильной или оксифильной цитоплазмой, для которой характерна раняя гемоглобинизация). В крови встречается много дегенеративно измененных эритроцитов:пойкилоцитоз, анизоцитоз с микроцитозом, гиперхромия, мегалоциты с патологическими включениями. Уменьшается количество клеток физиологической регенерации (ретикулоциты, полихроматофилы), т.к. в костном мозге наблюдается раздражение эритроцитарного ростка с преобладанием мегалобластического типа кроветворения над нормобластическим. Наблюдается тромбо — и лейкоцитопения с атипическими клетками.
Дефицит витамина B12 (цианкоболамина) > :
1. Нарушение перехода: фолиевая кислота > тетрагидрофолиевая кислота > тимин > ДНК. Нарушение клеточного деления, при котором страдают активно размножающиеся клетки:
а) кроветворной ткани (анемия);
б) ЖКТ (воспалительно-атрофические процессы в слизистой).
2. Нарушение перехода метилмалоновой кислоты в янтарную > накопление метилмалоновой кислоты, которая оказывает токсическое действие на нервную систему;
3. Синтез жирных кислот с измененной структурой > нарушение образования миелина.
Источник
В12-
и фолиеводефицитная анемия — анемия,
связанная с нарушением синтеза нуклеиновых
кислот и заменой эритробластического
типа кроветворения мегалобластическим
вследствие недостатка в организме
цианокобаламина (витамина В12)
и фолиевой кислоты (мегалобластная
анемия).
Этиология.
По этиологии эти анемии могут быть
приобретенными и наследственными.
Причины, общие для В12-
и фолиеводефицитной анемий, следующие:
недостаток этих
витаминов в пище (вскармливание грудных
детей козьим молоком, сухими молочными
смесями);нарушение всасывания
витаминов в тонкой кишке (при резекции
тощей кишки или поражении ее опухолью,
множественными дивертикулами, при
тропическом спру, дифиллоботриозе,
алкоголизме);повышенное расходование
витаминов при беременности (когда
эмбриональный тип кроветворения у
плода сменяется эритробластическим,
увеличивается потребление плодом
цианокобаламина и фолиевой кислоты
матери);нарушение депонирования
витаминов при диффузном поражении
печени (гепатит, цирроз). Кроме того,
дефицит цианокобаламина возникает в
результате нарушения образования
внутреннего фактора Касла — мукопротеида
(транскоррина) — при наследственном
дефекте выработки его клетками желез
желудка, при поражении слизистой
оболочки желудка опухолью, сифилитической
гуммой, большими дозами алкоголя, при
резекции желудка, разрушений мукопротеида
аутоантителами.
Причиной
возникновения пернициозной анемии
(злокачественной, анемии Аддисона —
Бирмера), являющейся разновидностью
В12-дефицитной
анемии, могут быть генетически
детерминированный дефицит транскоррина
(наследуется по аутосомно-рецессивному
типу) или же аутоиммунный процесс, о чем
свидетельствует обнаружение у больных
в сыворотке и желудочном соке антител
(IgG, IgA) к антигенам цитоплазмы париетальных
клеток, реже — к внутреннему фактору.
Патогенез.При
дефиците цианокобаламина (его кофермента
— метилкобаламина) не происходит
превращения фолиевой кислоты в ее
коферментную форму — тетрагидрофолиевую
кислоту, без которой невозможен синтез
тимидинмонофосфата, входящего в состав
ДНК. Нарушается клеточное деление и
прежде всего страдают активно
размножающиеся клетки кроветворной
ткани. В костном мозге задерживается
размножение и созревание эритрокариоцитов.,
эритробластический тип кроветворения
заменяется мегалобластическим, возрастает
неэффективный эритропоэз, укорачивается
продолжительность жизни эритроцитов.
Вследствие нарушения кроветворения и
гемолиза эритроцитов развивается
анемия, при которой клетки патологической
регенерации и эритроциты с признаками
дегенерации появляются не только в
костном мозге, но и в крови. Изменение
лейко- и тромбоцитопоэза проявляется
уменьшением числа лейкоцитов и
тромбоцитов, выраженной атипией клеток.
Возникновение
атипичного митоза и гигантских клеток
эпителия пищевого канала приводит к
развитию воспалительно-атрофических
процессов в слизистой оболочке его
отделов (глоссит, стоматит, эзофагит,
ахилический гастрит, энтерит). Это
усугубляет первичное нарушение секреции
и всасывания внутреннего фактора и,
следовательно, усиливает дефицит
витаминов (порочный круг).
В результате
недостатка цианокобаламина (его кофермент
дезоксиаденозилкобаламин участвует в
образовании янтарной кислоты из
метилмалоновой кислоты) в организме
накапливается метилмалоновая кислота,
токсичная для нервных клеток, а в нервных
волокнах синтезируются жирные кислоты
с измененной структурой, что отражается
на образовании миелина и ведет к
повреждению аксона. Развивается
дегенерация задних и боковых столбов
спинного мозга (фуникулярный
миелоз), поражаются
черепные и периферические нервы с
развитием многообразной неврологической
симптоматики.
Картина
крови. В12-
и фолиеводефицитные анемии — это анемии
с мегалобластическим типом кроветворения,
гиперхромные, макроцитарные. Содержание
эритроцитов и гемоглобина в крови при
этих анемиях может резко снижаться,
однако цветовой показатель выше 1
(1,4—1,8) в связи с наличием в крови больших
по объему мегалобластов и мегалоцитов,
насыщенных гемоглобином.
В мазке
крови появляются клетки патологической
регенерации костного мозга — мегалоциты
(интенсивно окрашенные клетки диаметром
10—12 мкм и более, не имеющие центрального
просветления, несколько овальной формы)
и единичные мегалобласты
(крупные клетки размером 12—15 мкм с
базофильной, полихроматофильной или
ацидофильной цитоплазмой и нежносетчатым,
обычно эксцентрично расположенным
ядром). Существует точка зрения, согласно
которой мегалобласты и мегалоциты при
В12-
и фолиеводефицитных анемиях не идентичны
эмбриональным клеткам эритроцитарного
ряда и только внешне похожи на них. В
крови встречается много дегенеративно
измененных эритроцитов: пойкилоцитоз,
анизоцитоз, гиперхромные мегало- и
макроциты, мегалоциты с включениями в
виде телец Жолли, колец Кебота, эритроциты
с базофильной зернистостью. Уменьшается
количество клеток физиологической
регенерации (ретикулоциты, полихроматофилы),
так как в костном мозге наблюдается
раздражение эритроцитарного ростка с
преобладанием мегалобластического
эритропоэза на фоне угнетения
эритробластического кроветворения.
Наблюдается лейко- и тромбоцитопения
с атипическими клетками (например,
гигантские полисегментированные
нейтрофильные гранулоциты размером
20—30 мкм, с 8—10 сегментами).
Соседние файлы в предмете Патологическая физиология
- #
- #
Источник
Мегалобластные
анемии
— группа заболеваний, хар-ся появлением
в красном костном мозге мегалобластов
— клеток красного ядра больших размеров
с измененной структурой ядра, которые
прослеживаются на всех стадиях
дифференцировки эритроидных
предшественников. Появление мегалобластов
связано с нарушением синтеза ДНК и
замедлением созревания клеток.
Витамин
B12-дефицитная
анемия
— группа заболеваний, связанных с
дефицитом цианокобаламина (витамин
B12)
или нарушением его метаболизма.
Причины
дефицита витамина В12:
1)
неполноценное питание (исключительно
вегетарианское);
2) дефицит внутреннего
фактора: гастрит типа А, иммунные антитела
к фактору Кастля или к париетальным
клеткам желудка, гастроэктомия, врожденный
дефицит его без нарушения функции
желудка, рак желудка;
3) хронический
панкреатит, синдром Золингера-Эллисона,
заболевания конечного отдела подвздошной
кишки (болезнь Крона, тяжелый энтерит),
синдром «приводящей кишки»;
4)
дисбактериоз (развитие бактерий в слепых
петлях);
5) наличие гельминтов (широкий
лентец);
6) хронический алкоголизм;
7)
заболевания печени (цирроз, гепатит);
8)
прием некоторых лекарственных средств,
избыточное применение антиметаболитов
при лечении опухолей также может вызвать
нарушение синтеза ДНК.
Клиника.
Болеют
лица в возрасте 45-65 лет, редко клиника
встречается у людей до 30 лет, чаще болеют
женщины. Обычно манифестация (прогрессия)
наступает весной или осенью. Заболевание
развивается постепенно, нередко долгое
время болезненное состояние проявляется
поражением ЖКТ, иногда признаками
поражения нервной системы (радикулиты),
адаптированной (легкой степени) анемией.
Выражены синдромы: поражение кроветворной
системы, ЖКТ и нервной системы. Клиника
проявляется слабостью, одышкой,
тахикардией, периодической диареей,
жгучими болями в языке при приеме острой
и кислой пищи, парестезиями, снижением
чувствительности, а при тяжелых анемиях
— спутанностью сознания, депрессией,
деменцией. Объективно: покровы бледные
с матовым и лимонно-желтым оттенком.
Язык гладкий, сглаженный, атрофичный
(иногда, гиперпластичный), блестящий,
влажный, иногда красный и воспаленный
(«лакированный» язык, глоссит Хантера).
Селезенка часто увеличена, иногда
увеличена печень. Парестезии пальцев
рук и ног. Атрофия мышц, полиневриты,
расстройства координации (неуверенная
или шаткая походка, нескоординированные
движения).
Диагностика:
— очень
низкое содержание витамина В12
в плазме (менее 100 пг/л, N 160-950 пг/л),
увеличение ферритина, уменьшение
гаптоглобина, повышение ЛДГ; — антитела
к внутреннему фактору или к париетальным
клеткам в сыворотке крови (обнаруживаются
в 50% случаев). — гиперхромия,
макроцитарная
гипо-, норморегенераторная (диспластическая)
анемия.
Обнаруживаются мегалоциты (11-14 мкм),
макроформы и анизоцитоз, включения в
эритроцитах (тельца Жолли, кольца Кебота,
зернистость Гейнца), полисегментация
нейтрофилов. — мегалобластный костный
мозг («синий» при окраске по Романовскому);
— отрицательный тест Шиллинга (при
экскреции мочи после приема радиоактивного
витамина В12
внутрь уменьшается выделение кобаламина);
— при Ph-метрии желудочного сока гипо- и
ахлоргидрия; — при биопсии слизистой
желудка — фундальный гастрит, гипертрофия
бокаловидных клеток, атрофия париетальных
и главных клеток, клеточный атипизм; —
увеличение билирубина за счет непрямого
(неконъюгированного).
Лечение.
Режим
амбулаторный или стационарный в
зависимости от тяжести анемии. Диета:
с повышенным содержанием белков,
преимущественно продукты животного
происхождения. Лекарственная терапия
заключается в назначении цианкобаламина
по 1000 мкг в/м 1 раз в день, далее по 500 мкг
3-4 недели, затем по 200 мкг/сут в течение
1-1,5 месяца или по 500 мкг 1 раз в неделю 1
месяц. С целью профилактики рецидивов
пожизненно вводят цианкобаламин
по 500 мкг 2 раза в месяц. Уже в первые
сутки после в/м введения витамина В12
происходит трансформация мегалобластного
типа в нормоцитарный, усиливается
эритропоэз. Через 48-72 часа после первой
инъекции витамина
В12начинает
увеличиваться число ретикулоцитов.
Ретикулоцитарный
криз нарастает
к 7-12 дню, что может быть оценено как
результат эффективной терапии. В
клинических условиях фолиевая кислота
не должна использоваться для лечения
В12-дефицитной
анемии, так как могут развиться серьезные
неврологические расстройства даже при
ликвидации анемии. Трансфузии эритроцитов
проводят при угрозе анемической комы
— наиболее грозного и плохо поддающегося
лечению осложнения В12-дефицитной
анемии. Прогноз
при адекватном лечении благоприятный.
Хронизация анемии отмечается при
алкоголизме и у лиц после тотальной
резекции желудка или кишечника. После
достижения ремиссии больные подлежат
диспансерному наблюдению, проведению
ФГС с биопсией слизистой желудка и
противорецидивному лечению витамином
В12
весной
и осенью.
Фолиеводефицитная
анемия
— анемия, обусловленная дефицитом
фолиевой кислоты или нарушения ее
утилизации в процессе эритропоэза, что
приводит к мегалобластному типу
кроветворения.
Патогенез.
Фолиевая
кислота
и ее соединения известны под названием
фолатов. Организм получает фолаты при
расщеплении содержащихся в пище
полиглютаматов в моноглютаматы в тонком
кишечнике. В плазме происходит превращение
метилтетрафолата в присутствии витамина
В12
в тетрагидрофолаты. Последние превращаются
в формил-ТГФ (фолиновая кислота), затем
в дезокситимидин-монофосфат с последующим
синтезирующим действием на ДНК. Таким
образом, дефицит витамина В12
ведет к нарушению синтеза фолиновой
кислоты и развитию В12-дефицитной
анемии. Дефицит фолиевой кислоты ведет
к развитию фолиеводефицитной анемии.
Суточная потребность фолатов составляет
100 мкг. Запасы, создаваемые в тканях
(печень), достаточны для синтезирования
ДНК в течение 1-3 мес.
Этиология.
1.
Недостаток фолиевой кислоты в пище (в
том числе вскармливание новорожденных
козьим или порошковым молоком). 2.
Нарушения всасывания в тонком кишечнике
и нарушения депонирования в печени (в
том числе при злоупотреблении алкоголем).
3. Прием антагонистов фолиевой кислоты
(метотрексата), аналогов пурина и
пиримидина, противосудорожных препаратов
— дифенина, фенобарбитала. 4. Повышение
потребности в фолиевой кислоте
(беременность, новорожденные,
миелопролиферативные синдромы,
хронический гемолиз). 5. При очень активной
пролиферации клеток (гемолиз, лейкозы
и другие опухоли, инфекции, псориаз).
6.
Угнетение системы дигидрофолат-редуктазы
(при приеме ЛС, при алкоголизме).
Клиника
сходна с В12-дефицитной
анемией. Не характерен атрофический
гастрит с ахилией, нет фуникулярного
миелоза. Не наблюдается геморрагический
диатез. Более выражены функциональные
признаки поражения ЦНС. Встречается
чаще у детей, молодых женщин, алкоголиков,
больных эпилепсией.
Диагностические
критерии дефицита фолиевой кислоты:
низкий уровень содержания фолатов в
сыворотке (натощак) 3-25 нг/мл; низкий
уровень содержания фолатов в эритроцитах
(N 100-415 нг/мл); макроцитарная диспластическая
анемия; мегалобластный костный мозг;
нормальный или несколько сниженный
уровень витамина В12
в сыворотке крови (менее 100 мг/мл, N
160-930 мг/мл).
Гемограмма
и миелограмма
сходны с витамин В12-дефицитной
анемией.
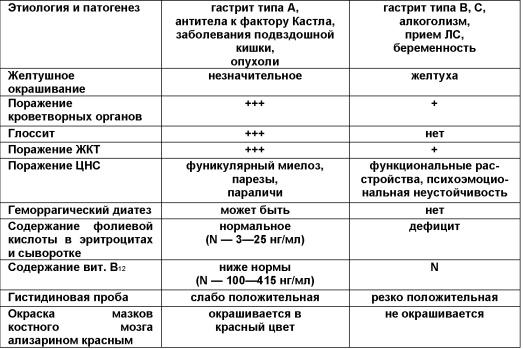
Дифференциальный
диагноз
с В12-дефицитной
анемией представлен в таблице.
Основные
дифференциально-диагностические
признаки витамин В12—
(2 столбик) и фолиеводефицитной анемий
(3 столбик)
Лечение.
Фолиевая
кислота внутрь
5-15 мг/сут на протяжении 4-6 недель до
получения ремиссии. В последующем при
неустраненной причине — поддерживающая
терапия 1-5 мг/сут.
40.
Хронический миелолейкоз: этиология,
патогенез, роль хромосомных аббераций
в развитии лейкоза, фазы лейкемического
процесса, терапия 1-ой линии, терапия
резерва. Показания к трансплантации
костного мозга, исходы и осложнения
ХМЛ.
Хронический
миелолейкоз (ХМЛ)
–
хронически протекающее миелопролиферативное
заболевание, при котором наблюдается
повышенное образование
гранулоцитов, преимущественно нейтрофилов,
являющихся субстратом опухоли. Источник
опухоли –
клетка-предшественник
миелопоэза.
Этиология.
Причиной
патологического роста клеток считается
мутация клетки-предшественника
миелопоэза (частично детерминированная
полипотентная клетка). Это доказывается
обнаружением у больных ХМЛ патологической
Ph-хромосомы
(филадельфийской) в клетках миелоидного,
эритроидного,
моноцитарного и тромбоцитарного рядов.
Ph-хромосома
является частым
клеточным маркером, подтверждающим
происхождение всего патологического
клона клеток при ХМЛ от одной материнской.
Несмотря на то, что лейкозными
являются все три ростка костного мозга,
в развернутой стадии ХМЛ
наблюдается безграничный рост, только
одного ростка –
гранулоцитарного. Существенно повышается
продукция и мегакариоцитов (тромбоцитов).
Классификация.
Заболевание закономерно проходит в
своем развитии две стадии –
моноклоновую (развернутая доброкачественная)
и поликлоновую (терминальную
злокачественную). Этому соответствуют
три фазы хронического миелолейкоза в
клиническом отображении:
—
хроническая –гиперплазия
гранулоцитарного ростка кроветворения
в красном костном мозге, при этом
способность клеток к дифференцировке
и созреванию сохранена; миелоидная
пролиферация костного мозга +
небольшие изменения
в крови без явлений интоксикации;
наблюдается умеренный лейкоцитоз, со
сдвигом лейкоцитарной формулы до
миелоцитов, увеличением содержания
зрелых и созревающих гранулоцитов в
красном костном мозге, эритро-
тромбоцитопоэз сохранены, селезенка
нормальных размеров;
—
фаза акселерации –
через 3-3,5 года выраженные
клинико-гематологические проявления
(интоксикация
продуктами распада лейкозных клеток,
увеличение печени и селезенки,
миелоидная пролиферация костного
мозга + изменения в крови: появляются
бластные клетки, анемия, тромбоцитопения;
нарастают симптомы интоксикации —
лихорадка, потливость, слабость, снижение
массы тела. Важный признак — резистентность
к успешно применявшейся ранее
химиотерапии);
—
бластный криз или терминальная
(соответствует
развитию поликлоновой опухоли) –
рефрактерность
к проводимой цитостатической терапии,
истощение, значительное увеличение
селезенки и печени, дистрофические
изменения внутренних органов,
выраженные изменения крови (анемия,
тромбоцитопения).
Появление
в периферической крови бластных клеток
(до 30–90%),
в связи с чем заболевание приобретает
черты острого лейкоза. Чаще
всего в костном мозге и периферической
крови бластный криз хар-ся
появлением миелобластов, однако могут
встретиться и недифференцируемые
бластные клетки. Одновременно
происходит значительное
угнетение тромбоцитопоэза, развивается
геморрагический синдром.
Клиническая
картина.
Миелопролиферативный
синдром (обусловлен
миелоидной пролиферацией костного
мозга) включает:
а)
общие
симптомы, вызванные интоксикацией,
разрастаниями лейкозных клеток
в костном мозге, селезенке и печени
(потливость, слабость, снижение массы
тела, тяжесть и боль в области селезенки
и печени), оссалгии.
б)
увеличение
печени и селезенки;
в)
лейкемические
инфильтраты в коже;
г)
характерные
изменения в костном мозге и
периферической крови.
Синдром,
обусловленный осложнениями:
а)
геморрагический
диатез (геморрагии и тромбозы вследствие
нарушения
прокоагулянтного и тромбоцитарного
звеньев гемостаза);
б)
гнойно-воспалительные
(пневмонии, плевриты, бронхиты, гнойные
поражения
кожи и подкожной клетчатки), обусловленные
резким снижением активности
иммунитета;
в)
мочекислый диатез (гиперурикемия
вследствие повышенного распада
гранулоцитов).
При
исследовании периферической
кровиобнаруживают:
лейкоцитоз
(кол-во
лейкоцитов колеблется в широких
пределах)
с появлением в лейкоцитарной формуле
пролиферирующих форм
(миелобласты и промиелоциты) и созревающих
гранулоцитов (миелоциты,
метамиелоциты). Функциональные
свойства лейкоцитов и содержание в них
ферментов изменены: снижена активность
щелочной фосфатазы нейтрофилов, нарушена
способность
к фагоцитозу.имеется
базофильно-эозинофильная ассоциация.в
ранних стадиях болезни
возможно обнаружение гипертромбоцитоза,
в дальнейшем — тромбоцитопения.развитие
нормоцитарной,
нормохромной анемии, связанной в
основном с вытеснением лейкозным клоном
красного
ростка кроветворения, можно наблюдать
в развернутой клинико-гематологической
стадии. В терминальной стадии анемия
становится еще более выраженной.ускорение
СОЭ
При
исследовании костного
мозга:
1) обнаруживают миелоидную пролиферацию
костного мозга, нормальный миелопоэз
полностью замещен патологическим
клоном. 2) В мазке костного мозга
преобладают гранулоциты: соотношение
лейкоциты/эритроциты достигает 10:1,
20:1
за счет увеличения гранулоцитов.
3) Если в периферической крови высокий
тромбоцитоз, в костном мозге
отмечается большое количество
мегакариоцитов.
При
пункции
увеличенной
селезенкиобнаруживается
преобладание миелоидных клеток.
Диагностическими
критериямизаболевания
являются:
лейкоцитоз
более 20 — 103
в 1 мкл крови;появление
в лейкоцитарной формуле пролиферирующих
форм (миелобласты и промиелоциты) и
созревающих гранулоцитов (миелоциты,
метамиелоциты);миелоидная
пролиферация костного мозга (по данным
миелограммы и
трепанобиопсии);снижение
активности щелочной фосфатазы нейтрофилов
(менее 25 ед);обнаружение
Ph-хромосомы
в кроветворных клетках;расширение
плацдарма кроветворения (по данным
сцинтиграфии костей);увеличение
размеров селезенки и печени.
ХМЛ
следует дифференцировать от так
называемых лейкемоидных реакций,
которые могут возникать при ряде
заболеваний (туберкулез, рак, различные
инфекции, почечная недостаточность и
пр.). По определению А.
И. Воробьева (1985), лейкемоидная
реакция
–
это «изменения в крови и органах
кроветворения, напоминающие лейкозы и
другие опухоли кроветворной системы,
но не трансформирующиеся в ту опухоль,
на которую они похожи». При лейкемоидной
реакции наблюдается высокий лейкоцитоз,
в периферической
крови появляются незрелые нейтрофилы,
однако базофильно-эозинофильная
ассоциация не обнаруживается.
Дифференциальный
диагноз
основывается
на выявлении основного заболевания
(рак, туберкулез и пр.), на
повышении активности щелочной фосфатазы
нейтрофилов (вместо ее снижения при
ХМЛ). При стернальной пункции для
лейкемоидной реакции характерно
увеличение содержания миелоцитов,
однако Ph-хромосома
никогда не
определяется.
Лечение.
Воздействие
на функционирование онкогена препаратом
иматиниб мезилат (торговое название —
Гливек) — ингибитор
ABL-тирозинкиназы.
Он соединяется с активными центрами
BCR-ABL-тирозинкиназы, что приводит к гибели
клеток, содержащих ее, т.е. Ph-положительных
клеток. Эффективность превосходит все
ранее известные терапевтические
средства, применяемые у больных ХМЛ
(миелосан, гидроксимочевина, интерферон-α,
аллотрансплатация). В настоящее время
во всем мире Гливек
является препаратом 1-й линии терапии.
Назначается по 400 мг в I стадии, по 600 мг
во II стадии, до 800 мг в III стадии курсами.
Аллотрансплантация гемопоэтических
стволовых клеток и препараты новой
генерации ингибиторов тирозинкиназ
(растительный алкалоид гомогаррингтоин,
который проявил высокую эффективность
в хронической фазе, в фазе акселерации
и даже в бластном кризе ХМЛ. Назначается
по 2,5 мг/кг в/в курсом до 14 дней, затем по
7 дней в месяц для поддержания ремиссии)
используются в качестве 2-й и последующих
линий терапии у больных в хронической
фазе ХМЛ с резистентностью к Гливеку
или его непереносимостью.
ПОКАЗАНИЯ
К ТРАНСПЛАНТАЦИИ КОСТНОГО МОЗГА:
—
Аллогенная
трансплантация костного мозга: острые
лейкозы; хронический миелолейкоз;
тяжелая апластическая анемия;
гемоглобинопатии; врожденные иммунодефициты
и нарушения метаболизма.
—
Аутологичная
трансплантация костного мозга:
злокачественные лимфомы; некоторые
солидные опухоли; аутоиммунные
заболевания.
Прогноз.
Длительность
жизни больных ХМЛ в среднем составляет
3–5
лет,
у отдельных больных достигает 10 лет и
более. Осложнения
ХМЛ. Острая
сердечно-сосудистая недостаточность,
инфекционные осложнения, ДВС-синдром
и др.
Профилактика.
Точных мер предупреждения ХМЛ не
существует, в связи с
чем можно говорить лишь о вторичной
профилактике болезни, которая состоит
в предупреждении обострений болезни
(поддерживающая терапия, исключение
инсоляции, простудных заболеваний).
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник
