Роль иммунного воспаления при атеросклерозе
Изучение воспаления сосудистой стенки поколебало господствовавшее в ХХ веке представление, будто атеросклероз обусловлен исключительно нарушением метаболизма отдельных фракций холестерина (ХС). Прогрессирование заболевания, особенно его осложненных форм (нестабильной стенокардии, инфаркта миокарда (ИМ), стали объяснять не только влиянием на эндотелий «злого ХС» — липопротеинов низкой плотности (ЛПНП), но и развитием в пораженной области сосудистой стенки воспаления, причем не всегда связанного с инфекционным возбудителем.
 Евгений АТРОЩЕНКО, зав. лабораторией хронической ИБС и сердечной недостаточности, доктор мед. наук;
Евгений АТРОЩЕНКО, зав. лабораторией хронической ИБС и сердечной недостаточности, доктор мед. наук;
 Ирэна КАРПОВА, ведущий научный сотрудник, канд. мед. наук, РНПЦ «Кардиология»
Ирэна КАРПОВА, ведущий научный сотрудник, канд. мед. наук, РНПЦ «Кардиология»
О чем «расскажет» СРБ
Реальность воспалительной теории подтверждается обнаружением в крови больных кардиопрофиля, в т. ч. коронарной болезнью сердца (КБС), повышенных маркеров системного воспалительного ответа — например, С-реактивного белка (СРБ). Первые убедительные данные, обосновывающие его значительную прогностическую роль как фактора осложнений атеросклероза сосудов, получены в исследовании MRFIT (Multiple Risk Faсtor Intervention Trial) в 1996 г. Показано: при высоком содержании в плазме крови СРБ риск ИМ и смерти от КБС в 3 раза выше, чем при нормальном. Дополнительные факторы усугубляют его: у курящих вероятность ИМ возрастает в 4 раза.
Обследования совершенно здоровых врачей-мужчин (Physicians Health Study) подтвердили: при высоком СРБ риск сердечно-сосудистых катастроф увеличивается в 2,9 раза. Близкие данные — в проспективном исследовании Helsinki Heart Study (2000), а также в MONIKA Augsburg Cohort Study (1999), в котором больные с перенесенным ИМ наблюдались 8 лет. В работе Women’s Health Study («Здоровье женщин») показано, что ассоциация высокого содержания СРБ и риска ИБС касается и представительниц слабого пола.
Эндотелий дает сбой
Чем можно объяснить столь тесную связь воспаления эндотелиального слоя артерий и атеросклероза? Эндотелий — функциональный барьер между стенкой сосуда и циркулирующей в нем кровью. Функциональный, поскольку работает как сложнейший орган весом в 5 сердец и площадью, эквивалентной 6 теннисным кортам (площадь сосудистой системы равна 1000 м2 и покрыта 1013 эндотелиальными клетками). Особенностью является то, что он рассредоточен по всему телу человека и играет огромную роль в гомеостазе, сосудистой активности, пролиферации клеток, иммунных реакциях и отклике на воспаление.
В норме эндотелий:
- обладает антикоагулянтной, фибринолитической и антитромботической способностями;
- участвует в метаболизме липопротеинов и эйкозаноидов;
- является избирательным барьером, препятствующим проникновению «тяжелых», высокомолекулярных белков и липопротеинов в окружающие сосуды ткани, что привело бы к блоку обмена и прекращению жизни;
- регулирует тонус гладкомышечных сосудов;
- участвует в росте сосудов, адгезии лейкоцитов и регуляции иммунного ответа на воздействие;
- защищает гладкие мышцы сосудов от действия сосудосуживающих провокаторов.
Как правило, при отсутствии артериальной гипертензии (АГ), сахарного диабета (СД), иных факторов повреждения эндотелий остается интактным в первые 10–20 лет жизни человека. К 30 годам формируется его дисфункция — различной степени выраженности. Проявляется снижением эндотелий-зависимой дилатации сосудов, виной чему нарушение обмена липидов — в сторону повышения продукции ХС ЛПНП. Последние подвергаются перекисному окислению, становятся более «агрессивными» и под воздействием СРБ усиленно поглощаются макрофагами. Прогрессирование атеросклероза сопровождается выработкой антител к модифицированным ЛПНП и образованием холестеринсодержащих иммунных комплексов (ИК). Помимо этого, модифицированные ЛПНП активируют клетки воспаления. А лейкоциты — провоспалительные цитокины: фактор некроза опухоли, интерлейкины (ИЛ) 1 и 2. ИЛ-1 контролирует синтез ИЛ-6, который индуцирует образование в печени СРБ. Последний не только накапливается в пенистых клетках атеросклеротических бляшек, но и откладывается в глубоком слое интимы сосудов, усиливая их повреждение и активизируя воспаление, поскольку связывается с комплексом комплемента. Т. е. асептическое воспаление сосудов поддерживается аутоиммунными нарушениями, что особенно характерно для осложненных форм атеросклероза.
Повышенный уровень СРБ ассоциируется со сниженной секрецией основного регулятора вазодилатации — оксида азота (NO) и повышенной продукцией мощного эндогенного вазоконстриктора эндотелина-1 (ЕТ-1), вырабатываемого эндотелиальными клетками, что еще более ухудшает функцию эндотелия.
На всех стадиях атеросклероза, и особенно при обострениях, наблюдается активация клеточного иммунитета как составляющего звена воспаления сосудов. При этом уже не имеет принципиального значения уровень ОХС, ХС ЛПНП и ЛПВП в плазме крови, поскольку иммуновоспалительный процесс запущен и будет прогрессировать. Поэтому по меньшей мере странной выглядит позиция тех врачей, которые начинают липидмодифицирующую терапию больных с КБС или ее эквивалентами (СД и пр.) только с этапа повышения уровня ОХС и/или ХС ЛПНП, а не с момента постановки диагноза атеросклеротического поражения одного или нескольких сосудистых бассейнов. В этом случае аргументом для терапии является не само заболевание, а одно из его проявлений — нарушение липидного обмена, служащее только «зеркалом» основной болезни, которую нужно лечить.
«Тушить пожар в бляшке» статинами
Поскольку самые ранние проявления атеросклероза начинаются с эндотелия, проницаемость которого для ХС ЛПНП повышается благодаря свободным радикалам О2 и увеличению продукции малональдегида, кажется логичным на этой доклинической стадии для профилактики назначать «антидоты» — например, такой известный, эндогенно вырабатываемый антиоксидант, как альфа-токоферол (витамин Е). Но последний не снижает уровень ОХС и ХС ЛПНП. У лиц с клиническими проявлениями атеросклероза он не оказывает существенного влияния на прогноз жизни пациентов. Антирадикальным действием обладает и триметазидин (в Беларуси — предуктал), что выражается в улучшении эндотелий-зависимой дилатации микрососудов.
Многие противовоспалительные лексредства — например, аспирин, применяемый даже в низких дозах (PHS), нестероидные противовоспалительные препараты и кортикостероиды (согласно исследованию MUNA) — способны снизить уровень СРБ. Но тот же метилпреднизолон (MUNA) ухудшил прогноз течения КБС, что не позволяет рекомендовать кортикостероиды для профилактики сердечно-сосудистых осложнений у больных с этим заболеванием.
Самыми надежными антивоспалительными средствами являются статины, что впервые установлено в 2 исследованиях по правастатину — CARE и PRINCE. Оказалось, эффективность этого класса препаратов основана не только на снижении уровня атерогенных липидов, но и на прямом противовоспалительном действии, позволяющем стабилизировать атеросклеротическую бляшку. Это особенно важно, когда она рыхлая, имеет тонкую «шапку» и готова к разрыву («уязвимая бляшка»), например, при нестабильной стенокардии.
Статины, как никакие другие лексредства, благотворно влияют на разные звенья процесса атеросклеротического поражения сосудов и, в первую очередь, — на воспаление в местах локализации «уязвимых бляшек», чем и объясняется высокая эффективность препаратов у пациентов с острым коронарным синдромом. Кратко суть их полезного действия в следующем. Статины снижают интенсивность миграции макрофагов и гладкомышечных клеток в сосудистой стенке, продукцию макрофагами металлопротеаз и противовоспалительных цитокинов, которые разрыхляют покрышку бляшки и увеличивают тем самым риск ее разрыва. Уменьшая выраженность оксидантного стресса и повышая продукцию NO, лекарства улучшают функцию эндотелия и убавляют риск тромбообразования. Вероятность тромбоза минимизируется не только за счет антивоспалительного влияния статинов непосредственно на бляшку, но и вследствие снижения уровня фибриногена в плазме крови, концентрации тканевого фактора плазминогена, угнетения продукции тромбоксана и нормализации АДФ-зависимой агрегации тромбоцитов. Еще в 2001 г. на сессии American College of Cardiology было доложено, что статины оказывают наиболее выраженное противовоспалительное действие у пациентов со стойким и выраженным повышением СРБ, особенно в сочетании с дислипопротеинемией. Их эффект проявляется спустя 12 недель от начала лечения, а первые противовоспалительные проявления — уменьшение интенсивности пролиферации и миграции гладкомышечных клеток — наступают уже через 6 дней.
Для рационального использования статинов как эффективных липидмодифицирующих и противовоспалительных лексредств, улучшающих функцию эндотелия, важно знать следующее. В кардиологии хорошо известен «синдром отмены», когда внезапное прекращение приема лекарства, например, бета-адреноблокатора, может привести к драматическим последствиям. Считалось, что на статины это правило не распространяется. Однако теперь установлено, что резкий отказ от статинов ведет к активации механизмов, значительно снижающих продукцию основного регулятора гомеостаза эндотелия — оксида азота. А это создает угрозу инфаркта миокарда, ухудшения течения КБС — из-за усиления риска локального тромбообразования и повышения количества вазоспастических реакций артерий сердца.
«Бойцы» — фибраты и иАПФ
Противовоспалительным эффектом воздействия на стенку сосудов обладают представители еще 2 классов препаратов, широко используемых в кардиопрактике. Как показали Фремингемское исследование и программа PROCAM, вне зависимости от уровня ХС ЛПНП, по мере повышения триглицеридов (ТГ) (более 200 мг/дл, или 2,3 ммоль/л) и снижения ХС ЛПВП (менее 40 мг/дл и, особенно, ниже 25 мг/дл) — риск КБС существенно возрастает.
В эксперименте на культуре клеток эндотелия еще в 90-х годах XX века Дж. Кокерия показал, что частицы ЛПВП способны подавлять индуцированную провоспалительными цитокинами продукцию молекул адгезии ICAM-1, VCAM и Е-селектина. Наиболее выраженным благоприятным влиянием на содержание ТГ и ХС ЛПВП оказывают фибраты, но единственным из них, обладающим противовоспалительным эффектом, является фенофибрат. Под его воздействием происходит уменьшение высвобождения медиаторов воспаления и апо-птоз («самоуничтожение») активированных макрофагов — этих главных виновников и активных участников воспалительных реакций в местах атеросклеротического поражения сосудистой стенки.
Ренин-ангиотензин-альдостеро-новая система (РААС) играет значительную роль в повреждении сосудов в случаях ее значительной активации, что наблюдается у многих пациентов кардиологической клиники. Препараты, способные снизить уровень активности «демонического» ангиотензина-2, уменьшают оксидантный стресс, продукцию эндотелина и альдостерона, скорость фиброза сосудистой стенки и склонность артерий к вазоконстрикции.
К таким лекарствам относят ингибиторы ангиотензинпревращающего фермента (иАПФ) и антагониста АТ1-рецепторов ангиотензина ІІ (АРА). В большей или меньшей мере вазопротекторным действием обладают все иАПФ, но в плане реального антисклеротического влияния у них нет класс-эффекта. Серьезная доказательная база, основанная на экспериментальном материале и данных клинических исследований, есть в отношении только 2 ингибиторов — периндоприла и рамиприла. В клинических условиях антисклеротическое действие этих лекарственных средств проявляется исключительно при их использовании в больших дозах: не менее 8 мг и 10 мг в сутки соответственно.
Таким образом, классические представления о лечении атеросклероза как исключительно о коррекции нарушений липидного обмена, сегодня дополнены пониманием необходимости воздействия на процесс воспаления сосудистой стенки, «тушения пожара» в уязвимых бляшках. Несмотря на установленную связь повышенной концентрации маркеров воспаления и сердечно-сосудистых катастроф, они должны расцениваться как не факторы риска, а показатели выраженности, агрессивности течения атеросклероза. Целесообразность определения СРБ для оценки вероятности развития осложнений ИБС, в т. ч. нестабильной стенокардии, рестенозов после операций по реваскуляризации миокарда, инфаркта миокарда, а также для определения эффективности антисклеротической и антивоспалительной терапии, сегодня не вызывает сомнений.
По меньшей мере странной выглядит позиция тех врачей, которые начинают липидмодифицирующую терапию больных с КБС или ее эквивалентами (СД и пр.) только с этапа повышения уровня ОХС и/или ХС ЛПНП, а не с момента постановки диагноза атеросклеротического поражения одного или нескольких сосудистых бассейнов. В этом случае аргументом для терапии является не само заболевание, а одно из его проявлений — нарушение липидного обмена, служащее только «зеркалом» основной болезни, которую нужно лечить.
Материал предназначен для врачей-кардиологов, терапевтов.
Источник
Совокупность экспериментальных и клинических данных, накопленных за последние 10 лет, позволила продемонстрировать фундаментальную роль воспаления в процессе атерогенеза. Пенистые клетки, которые образовались из макрофагов, проникших в артериальную стенку, служат резервуаром избытка липидов.
В сформировавшейся атеросклеротической бляшки (АБ) эти клетки кроме того, являются источником провоспалительных медиаторов (белков типа цитокинов и хемокинов и различных эйкозаноидов, а также липидов, например фактора активации тромбоцитов). Эти фагоцитирующие клетки способны также в больших количествах продуцировать в АБ различные окислители, например супероксид. В совокупности все эти медиаторы ускоряют в АБ воспалительный процесс и способствуют тем самым ее прогрессированию.
Термином «врожденный иммунитет» описывают именно этот тип усиления воспалительной реакции, не зависящей от стимуляции антигена.
Наряду с этим появляется все больше информации о существенном вкладе в прогрессирование АБ антигенспецифического иммунитета (приобретенного). Помимо мононуклеарных фагоцитов представлять антигены Т-клеткам могут дендритные клетки, составляющие немногочисленную, но крайне важную популяцию лейкоцитов в АБ.
К возможным антигенам, способным стимулировать приобретенный иммунный ответ, относятся модифицированные липопротеины, белки теплового шока, β2-гликопротеин Ib и возбудители инфекции. Ан-тигенпрезентирующие клетки (макрофаги, дендритные и эндотелиальные клетки) обеспечивают взаимодействие антигена с Т-клетками, при котором запускается их активация. Далее активированные Т-клетки секретируют огромное количество цитокинов, способных модулировать процесс атерогенеза.
Хелперные Т-клетки (Th), несущие на своей поверхности маркер CD4, представлены двумя категориями. Th подтипа 1 (Тh1) продуцируют провоспалительные цитокины, включая IFNy, лимфотоксин, лиганд CD40 и ФНОа. Цитокины, принадлежащие к подтипу ТЫ, активируют, в свою очередь, клетки сосудистой стенки и вызывают изменения биологии АБ, которые способны ее дестабилизировать и усилить тромбогенность.
В то же время хелперные Т-клетки, продуцирующие преимущественно цитокины подтипа 2 (Th2), например ИЛ-10, способны в ходе атерогенеза подавлять воспалительный процесс. Цитолитические Т-клетки (несущие маркер CD8) могут экспрессировать fas-лиганд и другие цитотоксические факторы, ускоряющие лизис и апоптоз клеток-мишеней, включая ГМК, ЭК и макрофаги. В месте атеросклеротического поражения может происходить гибель клеток всех трех указанных типов, что усугубляет его прогрессирование и осложнения.
Регуляторные Т-клетки способны продуцировать трансформирующий фактор роста β (transforming growth factor, TGFβ) и ИЛ-10. Регуляторные Т-лимфоциты экспрессируют маркеры CD4 и CD25. Оба соединения (TGFβ и ИЛ-10) обладают противовоспалительным эффектом. Результаты ряда экспериментов позволяют предполагать наличие антиатеросклеротической функции регуляторных Т-клеток в условиях in vivo. Роль В-клеток и антител в патогенезе атеросклероза остается до конца не изученной. Гуморальный иммунитет в зависимости от обстоятельств может быть атеропротективным или атерогенным.
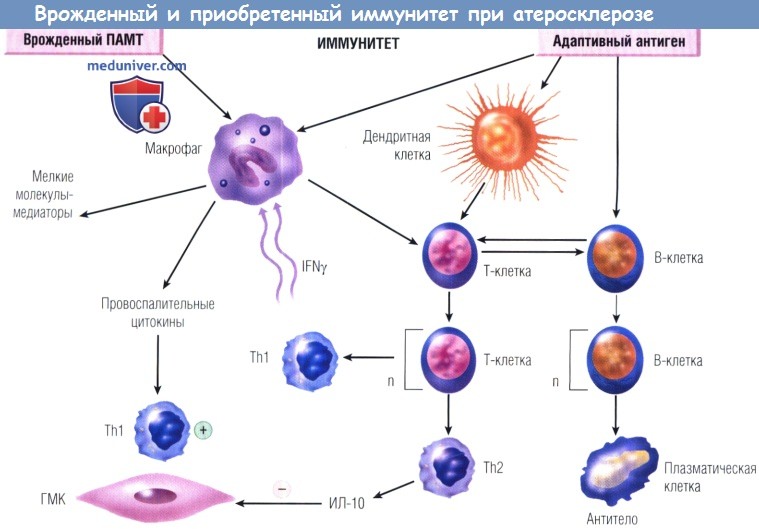
Врожденный и приобретенный иммунитет к атеросклерозу.
Представлена диаграмма путей действия врожденного (слева) и приобретенного (справа) иммунитета в процессе атерогенеза.
IFNy — интерферон гамма; Th — хелперная Т-клетка; ГМК — гладкомышечные клетки;
ИЛ-10 — интерлейкин 10; ПАМТ — патоген-ассоциированный молекулярный тип.
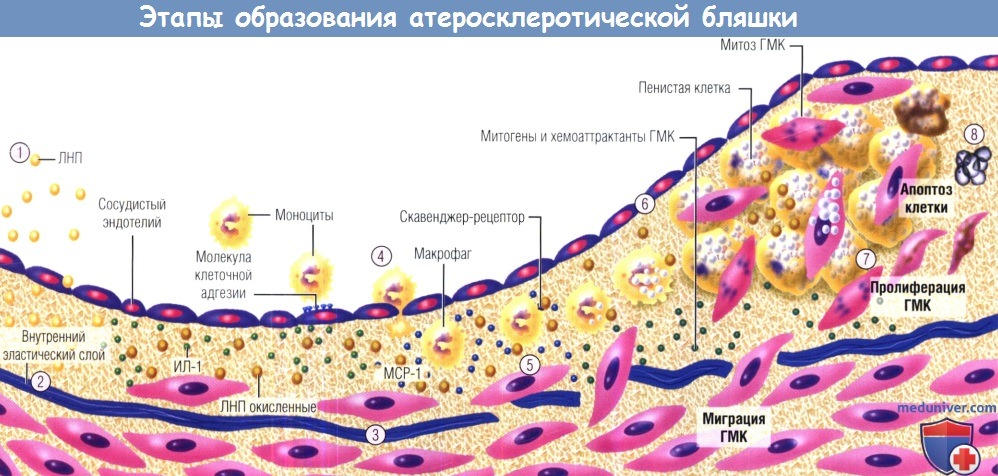
Схематическое изображение развития атеросклеротической бляшки.
Этап 1: накопление части липопротеинов в интиме; модифицированные липопротеины показаны более темным цветом, модификация — окисление и гликозилирование.
Этап 2: соединения, вызывающие окислительный стресс, включая модифицированные липопротеины, индуцируют локальную продукцию цитокинов.
Этап 3: цитокины, в свою очередь, индуцируют повышенную экспрессию молекул адгезии лейкоцитов, посредством которых последние прикрепляются к эндотелию, и хемоаттрактантов, которые обеспечивают их миграцию в интиму.
Этап 4: моноциты крови, попадающие в артериальную стенку за счет взаимодействия с цитокинами-хемоаттрактантами (например, МСР-1),
получают стимулы (например, от макрофагального колониестимулирующего фактора), усиливающие экспрессию ими скавенджер-рецепторов.
Этап 5: скавенджер-рецепторы опосредуют захват модифицированных липопротеинов и ускоряют образование пенистых клеток.
Образовавшиеся из макрофагов пенистые клетки служат источником медиаторов, включая другие цитокины и эффекторные молекулы, такие как гипохлорная кислота, супероксид (O2-) и матриксные металлопротеиназы.
Этап 6: ГМК интимы делятся, другие ГМК мигрируют из медии в интиму.
Этап 7: ГМК делятся и продуцируют внеклеточный матрикс, способствуя тем самым его накоплению в растущей атеросклеротической бляшке. В такой ситуации липидная полоса превращается в фиброзно-жировое отложение.
Этап 8: на последних этапах происходит кальцификация (не показана) и продолжается фиброз, сопровождаемый иногда гибелью ГМК (в т.ч. путем апоптоза).
В результате формируется почти бесклеточная фиброзная покрышка, окружающая богатое липидами ядро, в котором также могут содержаться гибнущие или мертвые клетки и их детрит (продукт их распада).
МСР-1 — моноритарный хемоаттрактантный белок 1; ГМК — гладкомышечные клетки; ИЛ-1 — интерлейкин 1; ЛНП — липопротеины низкой плотности.
— Читать «Миграция и пролиферация гладкомышечных клеток при атеросклерозе»
Оглавление темы «Атеросклероз и его осложнения»:
- Роль иммунитета в развитии атеросклероза
- Миграция и пролиферация гладкомышечных клеток при атеросклерозе
- Механизм гибели гладкомышечных клеток при атеросклерозе
- Внеклеточный матрикс артерии при атеросклерозе
- Рост сосудов в атеросклеротической бляшке — ангиогенез
- Отложение кальция в атеросклеротической бляшке — минерализация, кальцификация
- Сужение артерии (стеноз) как осложнение атеросклероза
- Тромбоз артерии как осложнение атеросклероза
- Воспаление атеросклеротической бляшки при атеросклерозе как причина осложнений
- Аневризмы артерий как осложнение атеросклероза
Источник
