Патогенетические механизмы развития атеросклероза
Клиническое значение атеросклероза и его осложнений пробудило огромный интерес к исследованию механизмов, лежащих в основе этого заболевания. Ранее были выдвинуты две основные гипотезы: одна из них подчеркивает значение пролиферации клеток интимы, другая фокусирует внимание на рецидивирующем тромбообразовании и организации тромбов.
Современные представления об атерогенезе заимствуют элементы обеих теорий и добавляют описанные ранее факторы риска. Согласно гипотезе «ответ на повреждение» атеросклероз представляет собой хроническую воспалительную реакцию и процесс заживления артериальной стенки в ответ на повреждение эндотелия. Поражение прогрессирует в результате взаимодействия модифицированных липопротеинов, макрофагов моноритарного происхождения и Т-лимфоцитов с нормальными клеточными компонентами артериальной стенки.
Соответственно этой модели атеросклероз обусловливают следующие патогенетические процессы:
— хроническое повреждение эндотелия приводит к повышенной сосудистой проницаемости, адгезии лейкоцитов и тромбозу;
— накопление липопротеинов (в основном ЛПНП и их окисленных форм) в стенке сосудов;
— адгезия моноцитов к эндотелию с их последующей миграцией и трансформацией в макрофаги и пенистые клетки;
— адгезия тромбоцитов;
— высвобождение факторов активированными тромбоцитами, макрофагами и клетками сосудистой стенки, включая миграцию гладкомышечных клеток либо из медии сосудистой стенки, либо из циркулирующих клеток-предшественников;
— пролиферация гладкомышечных клеток и образование ВКМ;
— накопление липидов как вне, так и внутри клеток (макрофагов и гладкомышечных клеток).
Теперь детально рассмотрим основные механизмы атерогенеза.
а) Повреждение эндотелия. В основе гипотезы «ответ на повреждение» лежит дисфункция эндотелия, а не утрата его вследствие любого типа повреждения (путем механического слущивания, под влиянием гемодинамических сил, отложения иммунных комплексов, облучения или химических веществ), когда происходит лишь утолщение интимы. Ранние повреждения стенки сосуда в случае богатой липидами диеты и других факторов риска появляются в участках морфологически интактного эндотелия, но с нарушенной функцией. Такой эндотелиальный слой имеет повышенную проницаемость, усиленную адгезию лейкоцитов и измененную экспрессию генов.
Специфические этиологические факторы, способствующие дисфункции эндотелия при раннем атеросклерозе, изучены недостаточно полно. К этим факторам относятся гипертензия, гиперлипидемия, токсичные вещества табачного дыма, гипергомоцистеинемия и инфекции. Воспалительные цитокины, в частности фактор некроза опухолей (TNF), также могут стимулировать проатерогенные формы экспрессии генов эндотелиальных клеток. Наиболее важные причины дисфункции эндотелия — гемодинамические нарушения и гиперхолестеринемия.
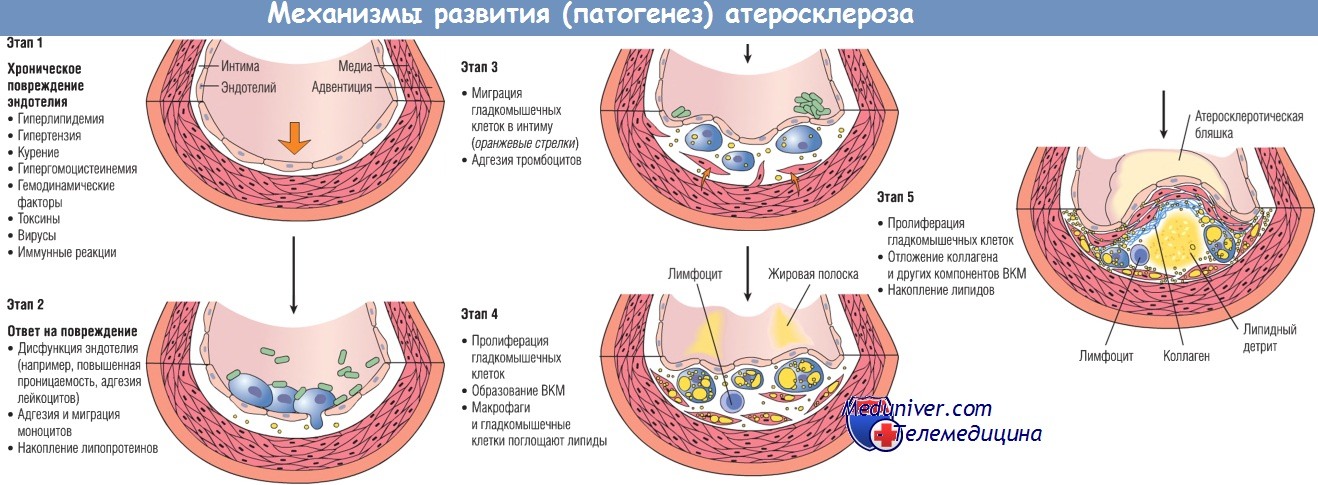
Развитие изменений артериальной стенки согласно гипотезе «ответ на повреждение».
ВКМ — внеклеточный матрикс.
б) Гемодинамические нарушения. Значение гемодинамических нарушений в патогенезе атерогенеза иллюстрирует факт, что бляшки имеют тенденцию локализоваться в устьях артерий крупного калибра, участках ветвления артерий и вдоль задней стенки брюшной аорты, т.е. там, где кровоток имеет особые характеристики. Исследования in vitro показали, что нетурбулентный ламинарный кровоток в нормальной сосудистой сети индуцирует гены, продукты которых (например, антиоксидантная супероксиддисмутаза) защищают от атеросклероза. Такие «атеропротективные» гены объясняют неслучайную локализацию ранних атеросклеротических поражений.
в) Липиды. Липиды обычно транспортируются с кровотоком, будучи связанными со специфическими апопротеинами (в виде липопротеиновых комплексов). Дислипопротеинемия является результатом мутаций, изменяющих апопротеины или клеточные рецепторы липопротеинов, а также результатом других состояний, влияющих на уровень липидов (например, нефротического синдрома, алкоголизма, гипотиреоза, сахарного диабета). К частым аномалиям липидного статуса (присутствующим у многих пациентов, перенесших инфаркт миокарда) относятся:
(1) повышенный уровень ЛПНП;
(2) сниженный уровень ЛПВП;
(3) повышенный уровень аномального липопротеина (а).
Причастность гиперхолестеринемии к атерогенезу доказывают следующие данные:
— преобладающими липидами в атеросклеротических бляшках являются холестерин и его эфиры;
— генетические дефекты захвата и метаболизма липопротеинов, обусловливающие липопротеинемию, ассоциируются с ранним атеросклерозом. Так, гомозиготная семейная гиперлипопротеинемия из-за дефекта рецепторов ЛПНП и неадекватного поглощения ЛПНП в печени может вызвать инфаркт миокарда до возраста 20 лет. Ранний атеросклероз получен в экспериментах на животных с генетически созданным дефицитом аполипопротеинов или рецепторов ЛПНП;
— другие генетические или приобретенные заболевания (например, сахарный диабет, гипотиреоз), вызывающие гиперхолестеринемию, способны стать причиной раннего атеросклероза;
— эпидемиологический анализ показал корреляцию между тяжестью атеросклероза и уровнем общего холестерина или ЛПНП в плазме;
— снижение уровня холестерина в сыворотке при соответствующей диете или на фоне приема лекарственных средств замедляет скорость прогрессирования атеросклероза, вызывает регрессию некоторых бляшек и снижает риск сердечнососудистых осложнений.
Механизмы, посредством которых хроническая гиперлипидемия способствует атерогенезу:
— хроническая гиперлипидемия, особенно гиперхолестеринемия, может непосредственно нарушать функцию эндотелиальных клеток, повышая местную продукцию свободных радикалов кислорода, называемых активными формами кислорода (АФК). АФК способны повреждать ткани за счет ускоренного распада оксида азота, что снижает его вазодилататорную активность;
— при хронической гиперлипидемии липопротеины накапливаются в интиме. Эти липиды окисляются под действием АФК, продуцируемых макрофагами или эндотелиальными клетками местно. Окисленные ЛПНП поглощаются макрофагами с помощью скавенджер-рецепторов, отличающихся от рецепторов ЛПНП, и накапливаются в фагоцитах (пенистых клетках). Кроме того, окисленные ЛПНП стимулируют высвобождение факторов роста, цитокинов и хемоки-нов эндотелиальными клетками и макрофагами, что усиливает миграцию моноцитов в поврежденные участки. Наконец, окисленные ЛПНП цитотоксичны для эндотелиальных и гладкомышечных клеток и способны индуцировать дисфункцию эндотелия. На роль окисленных ЛПНП в атерогенезе указывает факт их накопления в макрофагах на всех стадиях формирования атеросклеротических бляшек.
г) Воспаление. Процесс воспаления способствует инициации, прогрессированию и развитию осложнений атеросклероза. В норме сосудистая стенка не адгезирует воспалительные клетки, но уже на ранних стадиях атерогенеза артериальные эндотелиальные клетки с нарушенной функцией экспрессируют молекулы, стимулирующие адгезию лейкоцитов. В частности, молекула адгезии сосудистого эндотелия 1 (VCAM-1) обеспечивает адгезию моноцитов и Т-клеток к эндотелию. После этого они мигрируют в интиму под влиянием местно продуцируемых хемокинов.
Моноциты трансформируются в макрофаги и с высокой авидностью поглощают липопротеины, включая ЛПНП. Миграция и дифференцировка моноцитов в макрофаги (и в конечном итоге в пенистые клетки) теоретически носит протективный характер, поскольку эти клетки удаляют потенциально болезнетворные липидные частицы. Однако окисленные ЛПНП стимулируют активацию макрофагов и продукцию цитокинов (например, TNF). Это еще больше повышает адгезию лейкоцитов и продукцию хемокинов (например, моноцитарного хемоаттрактантного белка 1), что дает стимул для миграции дополнительного количества мононуклеарных воспалительных клеток. Активированные макрофаги продуцируют также реактивные метаболиты кислорода, усиливающие окисление ЛПНП, и секретируют факторы роста, индуцирующие пролиферацию гладкомышечных клеток.
Т-лимфоциты, привлеченные в интиму, взаимодействуют с макрофагами и поддерживают хроническую воспалительную реакцию. Нет четкого представления, реагируют ли Т-клетки на специфические антигены (например, на бактериальные или вирусные антигены, на белки теплового шока, на модифицированные составляющие сосудистой стенки и липопротеины) или они неспецифически активируются в условиях локального воспаления. Как бы там ни было, активированные Т-клетки вырабатывают в зоне повреждения интимы воспалительные цитокины (например, IFN-y), которые могут стимулировать макрофаги, эндотелиальные и гладкомышечные клетки.
Вследствие хронического воспаления активированные лейкоциты и клетки сосудистой стенки высвобождают факторы роста, стимулирующие пролиферацию гладкомышечных клеток и синтез ВКМ.
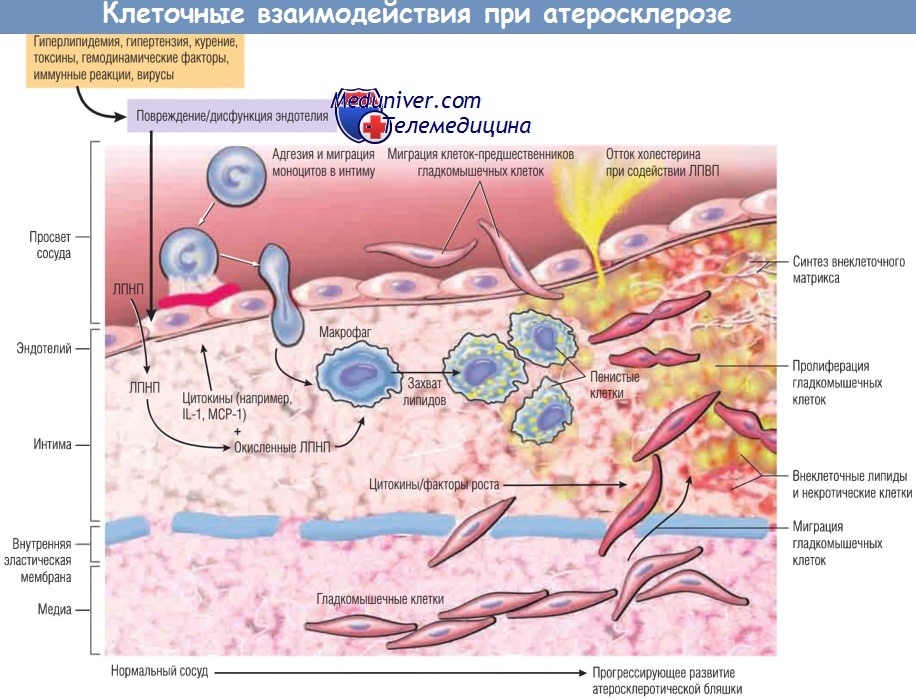
Гипотеза клеточных взаимодействий при атеросклерозе.
Предполагается, что гиперлипидемия и другие факторы риска повреждают эндотелий,
что приводит к адгезии тромбоцитов и моноцитов и высвобождению факторов роста (включая тромбоцитарный фактор роста),
индуцирующих миграцию и пролиферацию гладкомышечных клеток.
Пенистые клетки атеросклеротических бляшек образуются из макрофагов с помощью рецепторов липопротеинов очень низкой плотности и скавенджер-рецепторов модифицированных липопротеинов низкой плотности (ЛПНП),
например окисленных ЛПНП, и гладкомышечных клеток (механизм образования менее изучен).
Внеклеточные липиды попадают в бляшку из просвета сосуда, особенно в случае гиперхолестеринемии, а также из дегенерирующих пенистых клеток.
Накопление холестерина в бляшке отражает дисбаланс между притоком и оттоком холестерина,
а липопротеины высокой плотности (ЛПВП), вероятно, способствуют выведению холестерина из бляшки.
Гладкомышечные клетки мигрируют в интиму, пролиферируют и продуцируют ВКМ, включая коллаген и протеогликаны.
IL-1 — интерлейкин-1; МСР-1 — моноритарный хемоаттрактантный белок 1.
д) Инфекция. Было бы заманчиво предположить, что инфекция может индуцировать местный воспалительный процесс, лежащий в основе атеросклероза, однако эта гипотеза еще нуждается в доказательстве. В атеросклеротических бляшках обнаружены герпес-вирус, цитомегаловирус и С. pneumoniae, и у пациентов с тяжелым атеросклерозом сероэпидемиологические данные указывают на повышенный титр антител к С.pneumoniae. Не ясно, участвует ли С. pneumoniae в патогенезе атеросклероза, поскольку она часто ассоциируется с бронхитом и курением, которое является фактором риска ИБС. Данная инфекция широко распространена (как и атеросклероз), поэтому трудно определить, есть ли причинная связь или это просто совпадение.
Тем не менее вполне возможно, что микроорганизмы способны инфицировать атеросклеротические бляшки на ранней стадии их формирования, при этом чужеродные антигены могут потенцировать атерогенез, индуцируя местный иммунный ответ, или инфекционный агент способствует развитию местного протромботического статуса.
и) Пролиферация гладкомышечных клеток. Пролиферация гладкомышечных клеток и формирование ВКМ превращают жировые полоски (наиболее ранняя стадия поражения) в зрелую атеросклеротическую бляшку и способствуют ее увеличению. (Напомним, что гладкомышечные клетки интимы могут быть производными клеток-предшественников, циркулирующих в крови, и характеризуются пролиферативным и синтетическим фенотипами, что отличает эти клетки от гладкомышечных клеток подлежащей медии.) В пролиферации этих клеток и синтезе ВКМ участвуют некоторые факторы роста, включая PDGF (высвобождаемый локально прилипшими тромбоцитами, а также макрофагами, эндотелиальными и гладкомышечными клетками), фактор роста фибробластов и трансформирующий фактор роста a (TGF-a).
Мигрировавшие гладкомышечные клетки синтезируют ВКМ (в т.ч. коллаген), стабилизирующий атеросклеротические бляшки, но активированные воспалительные клетки в атеросклеротической бляшке могут вызвать апоптоз гладкомышечных клеток интимы, а также повысить катаболизм ВКМ, приводя к нестабильности бляшек.
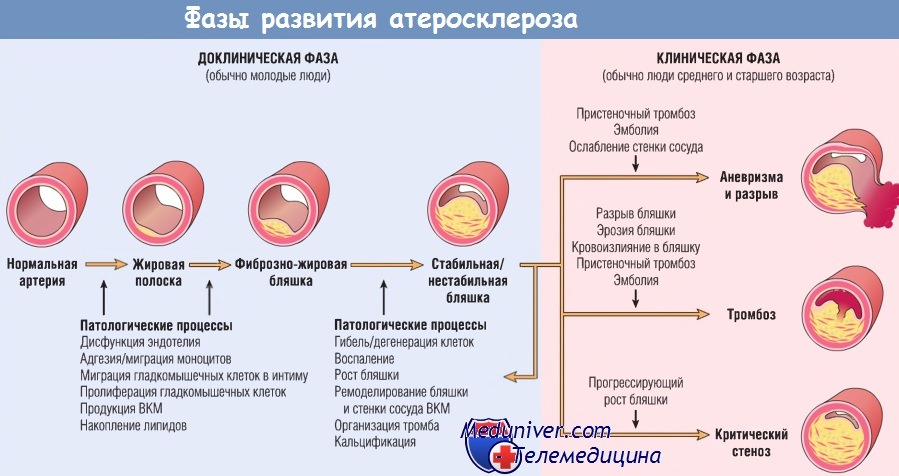
На рисунке ниже проиллюстрирована гипотеза развития атеросклероза как хронической воспалительной реакции (вместе с процессами, направленными в конечном итоге на попытку сосудистого «исцеления»), индуцированной разнообразными воздействиями, включая повреждение эндотелия, накопление и окисление липидов и тромбоз. Атеросклеротические бляшки — патологические динамические образования, состоящие из эндотелиальных клеток с нарушенной функцией, мигрировавших и пролиферирующих гладкомышечных клеток с примесью лимфоцитов и макрофагов. Все эти клетки способны секретировать медиаторы, влияющие на атерогенез.
Таким образом, бляшки в интиме на ранних стадиях развития немного больше по размеру, чем агрегаты, состоящие из гладкомышечных и макрофагальных пенистых клеток. По мере прогрессирования патологического процесса атеросклеротическая бляшка видоизменяется за счет ВКМ, синтезируемого гладкомышечными клетками. В интиме особенно хорошо выражена соединительная ткань, образующая фиброзную покрышку бляшки, при этом поражение обычно сохраняет центральное ядро из нагруженных липидами клеток и липидного детрита, который может кальцифицироваться. Со временем бляшка в интиме все больше выступает в просвет сосуда или сдавливает подлежащую медию, вызывая ее дегенерацию. Разрыв фиброзной покрышки может привести к тромбозу и острой окллюзии сосуда.
— Рекомендуем ознакомиться со следующей статьей «Морфология атеросклероза»
Оглавление темы «Атеросклероз и его последствия»:
- Причины и факторы риска атеросклероза
- Механизмы развития (патогенез) атеросклероза
- Морфология атеросклероза
- Последствия и осложнения атеросклероза
- Механизмы развития аневризмы и ее расслоения
- Механизмы развития и морфология аневризмы брюшной аорты
- Признаки аневризмы брюшной аорты
- Признаки аневризмы грудной аорты
- Механизмы развития (патогенез) расслоения аорты
- Морфология расслоения аорты
Источник
Атеросклероз
– это
хроническое заболевание сосудов, которое
характеризуется образованием в стенках
сосудов атеросклеротических бляшек
(отложение жиров и разрастание
соединительной ткани), которые сужают
и деформируют сосуды, что в свою очередь
является причиной нарушений циркуляции
крови и поражению внутренних органов.
этиологические
факторы: частые и длительные стрессы,
злоупотребление жирной и богатой
рафинированными углеводами пищей,
эндокринные и обменные заболевания
(ожирение, сахарный диабет, гипотиреоз,
желчно-каменная болезнь).
Основные
факторы риска развития атеросклероза:
артериальная гипертензия, повышение
вязкости и свертываемости крови,
дислипопротеинемия с повышением
содержания в сыворотке крови липопротеинов
очень низкой и низкой плотности (II-IV тип
гиперлипопротеинемии по классификации
ВОЗ) и снижением содержания липопротеинов
высокой плотности (ЛВП), курение, ожирение,
генетическая предрасположенность к
преждевременному атеросклерозу,
гиподинамия, возраст старше 40 лет.
Выделяют
5 стадий этого заболевания:
1
стадия — долипидная стадия: в этой стадии
нарушается проницаемость внутренней
оболочки сосудов для липопротеинов.2
стадия — стадия липоидоза: на внутренней
оболочке сосудов начинают накапливаться
липопротеины, они приобретают вид
желтоватых полосок.3
стадия — стадия липосклероза: на этой
стадии вокруг жиробелковых комплексов
разрастается соединительная ткань,
вследствие чего формируются выступающие
бляшки.4
стадия — стадия атероматоза: центр
бляшки начинает изъязвляться, она
значительно выступает в просвет сосуда,
под бляшкой начинает разрушаться слой
сосудистой стенки и она проникает в
мышечный слой сосуда.5
стадия — стадия атерокальциноза: на
этой стадии в бляшку происходит отложение
солей кальция, бляшка становится очень
плотной и значительно суживает просвет
сосуда, препятствуя току крови.
Патогенез
атеросклероза
Патогенез
атеросклероза сложен. По современным
представлениям в основе возникновения
атеросклероза лежит взаимодействие
многих патогенетических факторов,
ведущее в конечном счете к образованию
фиброзной бляшки (неосложненной и
осложненной).
Различают
три основные стадии формирования
атеросклеротической бляшки (атерогенез):
1. Образование
липидных пятен и полосок (стадия
липоидоза).
2. Образование
фиброзной бляшки (стадия липосклероза).
3. Формирование
осложненной атеросклеротической бляшки.
Начальная
стадия характеризуется появлением в
интиме артерий пятен и полосок, содержащих
липиды.
Атеросклероз, последствия которого
приводят к развитию ишемической
болезни сердца (ИБС), является одной
из самых частых причин смерти взрослого
населения в большинстве развитых
стран мира.(инсульт,инфаркт, стенокардия)
Лекарственная
терапия направлена
на контроль и воздействие на уровень
сывороточного холестерина, ли-попротеидов,
а также на питание и укрепление сосудистой
стенки. С целью снижения содержания
холестерина в крови больных атеросклерозом
используют много лекарственных препаратов
(клофибрат, полиспонин, никотиновую
кислоту, холестирамин и другие). Для ‘
воздействия на сосудистую стенку
назначают пармидин (ангинин, продектин).
Однако их недостаток в том, что
нормализующее действие проявляется
только в период приема препарата и
исчезает после отмены, отсюда необходимость
в длительном применении — месяцы, годы,
что не всегда удобно и желательно для
больного, ведь химиопрепараты далеко
не безвредны для организма, поэтому
особую значимость в лечении атеросклероза
приобретают лекарственные средства,
приготовленные из растительного сырья.
Среди них наибольшее-применение нашли
лекарственные травы и препараты из них,
влияющие на холестериновый обмен. В
частности, полиспонин — сухой экстракт
(таблетки) из корневищ и корней диоскореи
ниппонской, который назначается по
0,1—0,2 3 раза в день после еды в течение
месяца, курс лечения повторяется 3—4
раза через 7—10-дневный перерыв.
Соседние файлы в предмете Патологическая физиология
- #
- #
- #
- #
Источник
Атеросклероз
является полиэтиологическим заболеванием, в возникновении которого играют роль многочисленные факторы риска.
Патогенез атеросклероза сложен. По современным представлениям в основе возникновения атеросклероза лежит взаимодействие многих патогенетических факторов, ведущее в конечном счете к образованию фиброзной бляшки (неосложненной и осложненной).
Различают три основные стадии формирования атеросклеротической бляшки (атерогенез):
- Образование липидных пятен и полосок (стадия липоидоза).
- Образование фиброзной бляшки (стадия липосклероза).
- Формирование осложненной атеросклеротической бляшки.
Начальная стадия характеризуется появлением в интиме артерий пятен и полосок, содержащих липиды.
- Образование липидных пятен и полосок
Липидные пятна представляют собой небольших размеров (до 1,0-1,5 мм) участки на поверхности аорты и крупных артерий, которые имеют желтоватый цвет. Липидные пятна состоят, главным образом, из пенистых клеток, содержащих большое количество липидов и Т-лимфоцитов. В меньшем количестве в них присутствуют также макрофаги и гладкомышечные клетки. Со временем липидные пятна увеличиваются в размерах, сливаются друг с другом и образуют так называемые липидные полоски, слегка возвышающиеся надо поверхностью эндотелия. Они также состоят из макрофагов, лимфоцитов, гладкомышечных и пенистых клеток, содержащих липиды. На этой стадии развития атеросклероза
холестерин
расположен преимущественно внутриклеточно и лишь небольшое его количество находится вне клеток.

Липидные пятна и полоски образуются в результате отложения липидов в интиме артерий. Первым звеном этого процесса является повреждение эндотелия и возникновение эндотелиальной дисфункции, сопровождающееся повышением проницаемости этого барьера.
Причинами первоначального повреждения эндотелия могут служить несколько факторов:
- Механическое воздействие на эндотелий турбулентного потока крови, особенно в местах разветвления артерий.
- Артериальная гипертензия, увеличивающая напряжение сдвига.
- Увеличение в крови атерогенных фракций
ЛПНП
и
липопротеина (а)
, особенно их модифицированных форм, образующихся в результате перекисного окисления липидов или их гликозилирования (при сахарном диабете) и обладающие выраженным цитотоксическим действием.
- Повышение активности симпато-адреналовой и ренин-ангиотензиновой систем, сопровождающееся цитотоксическим действием катехоламинов и ангиотензина II на сосудистый эндотелий.
- Хроническая гипоксия и гипоксемия любого происхождения.
- Курение.
- Повышение у в крови содержания
гомоцистеина
, например, при дефиците витамина В
6
, В
12
и
фолиевой кислоты
.
- Вирусная и хламидийная инфекция, сопровождающаяся развитием хронического воспаления в стенке артерии.
В результате повреждения эндотелия формируется эндотелиальная дисфункция, проявляющаяся снижением продукции вазодилатирующих факторов (простациклин, окись азота и др.) и увеличением образования вазоконстрикторных веществ (эндотелинов, АII, тромбоксана А2 и др.), еще больше повреждающих эндотелий и повышающих его проницаемость. Модифицированные
ЛПНП
и
липопротеин (а)
и некоторые клеточные элементы крови (моноциты, лимфоциты) проникают в интиму артерий и подвергаются окислению или гликозилированию (модификации), что способствует еще большему повреждению эндотелия и облегчает миграцию из кровотока в интиму артерий этих клеточных элементов.

Моноциты, проникшие в интиму, трансформируются в макрофаги, которые с помощью так называемых скэвеннджер-рецепторов («рецепторов-мусорщиков») поглощают модифицированные
ЛПНП
и накапливают свободный и этерифицированный
холестерин
. Перегруженные липидами макрофаги превращаются в пенистые клетки. Макрофаги, перегруженные модифицированными ЛПНП, а также тромбоциты, проникающие в интиму артерий из крови, секретируют факторы роста и митогены, воздействующие на гладкомышечные клетки, расположенные в средней оболочке артерий. Под действием факторов роста и митогенов гладкомышечные клетки мигрируют в интиму и начинают пролиферировать. Находясь в интиме, они захватывают и накапливают модифицированные ЛПНП, также превращаясь в своеобразные пенистые клетки. Кроме того, гладкомышечные клетки приобретают способность сами продуцировать элементы соединительной ткани (коллаген, эластин, и гликозамингликаны), которые в дальнейшем используются для построения фиброзного каркаса атеросклеротической бляшки. Со временем пенистые клетки подвергаются апоптозу. В результате липиды попадают во внеклеточное пространство.
Липидные пятна появляются в артериях с раннего детства. В возрасте 10 лет липидные пятна занимают около 10% поверхности аорты, а к 25 годам — от 30 до 50% поверхности. В венечных артериях сердца липоидоз встречается с 10-15 лет, а в артериях мозга — к 35-45 годам.
- Образование фиброзных бляшек
По мере прогрессирования патологического процесса в участках отложения липидов разрастается молодая соединительная ткань, что ведет к образованию фиброзных бляшек, в центре которых формируется так называемое липидное ядро. Этому способствует увеличение количества липидов, высвобождающихся в результате гибели (апоптоза) гладкомышечных клеток, макрофагов и пенистых клеток, перегруженных липидами. Экстрацеллюлярно расположенные липиды пропитывают интиму, образуя липидное ядро, которое представляет собой скопление атероматозных масс (липидно-белкового детрита). Вокруг липидного ядра возникает зона соединительной ткани, которая на начальном этапе богата клеточными элементами (макрофагами, пенистыми и гладкомышечными клетками, Т-лимфоцитами), коллагеном и эластическими волокнами.
Одновременно происходит васкуляризация очага атеросклеротического поражения. Вновь образующиеся сосуды отличаются повышенной проницаемостью и склонностью к образованию микротромбов и разрывам сосудистой стенки. По мере созревания соединительной ткани количество клеточных элементов уменьшается, а коллагеновые волокна утолщаются, формируя соединительнотканный каркас атеросклеротической бляшки, который отделяет липидное ядро от просвета сосуда («покрышка»). Формируется типичная фиброзная бляшка, выступающая в просвет сосуда и нарушающая кровоток в нем.
Клиническое и прогностическое значение сформировавшейся атеросклеротической бляшки во многом зависит именно от структуры ее фиброзной покрышки и размеров липидного ядра. В некоторых случаях (в том числе на относительно ранних стадиях формирования бляшки) ее липидное ядро хорошо выражено, а соединительнотканная капсула сравнительно тонкая и может легко повреждаться под действием высокого артериального давления, ускорения кровотока в артерии и других факторов. Такие мягкие и эластичные бляшки иногда называют «желтыми бляшками». Они, как правило, мало суживают просвет сосуда, но ассоциируются с высоким риском возникновения повреждений и разрывов фиброзной капсулы, то есть с формированием так называемой «осложненной» атеросклеротической бляшки.
В других случаях (обычно на более поздних стадиях) фиброзная покрышка хорошо выражена, плотная и меньше подвержена повреждению и разрывам. Такие бляшки называют «белыми». Они нередко значительно выступают в просвет артерии и вызывают гемодинамически значимое ее сужение, которое в некоторых случаях может осложняться возникновением пристеночного тромба.
Первые две стадии атерогенеза завершаются образованием неосложненной атеросклеротической бляшки. Прогрессирование атероматозного процесса приводит к формированию «осложненной» атеросклеротической бляшки, вследствие чего образуется пристеночный тромб, который может приводить к внезапному и резкому ограничению кровотока в артерии.

- Формирование «осложненной» бляшки
Прогрессирование атероматозного процесса приводит к формированию «осложненной» атеросклеротической бляшки. Эта стадия атероматоза характеризуется значительным увеличением липидного ядра (до 30% и более от общего объема бляшки), возникновением кровоизлияний в бляшку, истончением ее фиброзной капсулы и разрушением покрышки с образованием трещин, разрывов и атероматозных язв. Выпадающий при этом в просвет сосудов детрит может стать источником эмболии, а сама атероматозная язва служить основой для образования тромбов. Завершающей стадией атеросклероза является атерокальциноз, отложение солей кальция в атероматозных массы, межуточное вещество и фиброзную ткань.

На рисунке схематично представлены все три стадии формирования атеросклеротической бляшки.
Главным следствием формирования «осложненной» атеросклеротической бляшки является образование пристеночного тромба, который внезапно и резко ограничивает кровоток в артерии. В большинстве случаев именно в этот период возникают клинические проявления обострения заболевания, соответствующие локализации атеросклеротической бляшки (нестабильная стенокардия, инфаркт миокарда, ишемический инсульт и т.п.).
Таким образом, наиболее значимыми осложнениями атеросклеротического процесса являются:
- Гемодинамически значимое сужение просвета артерии за счет выступающей в просвет артерии атеросклеротической бляшки.
- Разрушение фиброзной капсулы, ее изъязвление, что способствует агрегации тромбоцитов и возникновению пристеночного тромба.
- Разрыв фиброзной капсулы атеросклеротической бляшки и выпадение в просвет сосуда содержимого липидного ядра детрита, который может стать источником эмболии и ли формирования пристеночного тромба.
- Кровоизлияние в бляшку из вновь образованных микрососудов, что также способствует разрыву покрышки и формированию тромба на поверхности атеросклеротической бляшки и т.д..
- Отложение солей кальция в атероматозные массы, межуточное вещество и фиброзную ткань, что существенно увеличивает плотность атеросклеротической бляшки.
- Стабильные и нестабильные атеросклеротические бляшки
Атеросклеротические бляшки могут быть стабильными и нестабильными.
Стабильность бляшки зависит от ее строения, размеров и конфигурации. Стабильные бляшки статичны или характеризуются медленным ростом в течение многих лет. Стабильные бляшки богаты коллагеном; нестабильные — липидами.

Тонкая фиброзная капсула атеросклеротической бляшки (между стрелками) отделяет мягкое липидное ядро от просвета сосуда.

Тонкая фиброзная капсула, инфильтрированная макрофагами (пенистыми клетками), покрывающая липидное ядро бляшки, способна к разрыву. Если на поверхности такой капсулы имеются эритроциты, вероятность ее разрыва очень высока.
Нестабильные бляшки легко подвергаются эрозии, разрывам, приводя к острым тромбозам, окклюзиям и инфарктам еще до развития стенозов сосудов.

Тонкая, инфильтрированная пенистыми клетками капсула бляшки (между стрелками) в состоянии разрыва.

D — разрыв (обозначен стрелками) тонкой, инфильтрированной пенистыми клетками капсулы бляшки, пристеночный тромбоз в месте разрыва.
Клинические признаки атеросклероза появляются при прогрессирующем сужении просвета артерии в результате разрастания стабильной атеросклеротической бляшки, когда дефициту кровотока составляет 50-70%. В этом случае развиваются:
- Cтабильная стенокардия.
- Перемежающаяся хромота.
- Мезентериальная стенокардия и другие проявления атеросклероза.

На рисунке схематично представлен механизм формирования нестабильной бляшки.
Разрыв нестабильной атеросклеротической бляшки и оголение эндотелия ведут к образованию тромба, который полностью или частично препятствует кровотоку в пораженной артерии. При этом диагностируются:
- Нестабильная стенокардия.
- Инфаркты миокарда.
- Транзиторные ишемические атаки.
- Инсульты.
Атероматозная эмболия возникает спонтанно:
- Как осложнение хирургических операций на аорте.
- При проведении ангиографии, тромболитической терапии у пациентов с диффузным или прогрессирующим атеросклерозом.
(1).jpg)
На серии изображений, полученных в ходе микроскопии представлено атеросклеротическое поражение коронарных артерий, которое привело к их полной окклюзии:
- А — степень выраженности стеноза 90%.
- В, С — небольшие каналы реканализации в капиллярах.
- D — воспалительный процесс в адвентициальном слое сосуда.
- Е -разрастание капилляров в адвентициальной оболочке сосудов.
Источник

