Гемоглобинопатии талассемия и серповидно клеточная анемия
Гемоглобинопатии
преимущественно поражают население
тропических и субтропических областей
(Экваториальная Африка, Аравийский
полуостров, Южная Индия, Южный Китай,
Средиземноморье, Азербайджан, Грузия).Наиболеераспространены
и отличаются тяжестью проявлений
серповидноклеточная анемия и
большая талассемия, или анемия Кули.
Большинство гемоглобинопатий клинически
не проявляются; при некоторых — могут
наблюдаться: анемия, эритроцитоз или
цианоз (например, при метгемоглобинопатиях).
а)
Нарушения первичной структуры глобиновых
цепей гемоглобина вследствие
генной мутации. Описано около 500 аномальных
гемоглобинов.
Серповидно-клеточная анемия (ска)
Тип
наследования аутосомно-рецессивный,
дефект бета-глобиновых цепей, в которых
в 6 положении гидрофильный глютамин
заменен гидрофобным валином, образуется
HbS.
Восстановленная
форма HbS
мало растворима. При гипоксемии и
снижении скорости кровотока HbSполимеризуется
в длинные нерастворимые нити,
растягивающие эритроциты в форме серпа.
Если содержание HbS
больше 45% (гомозиготное состояние),
образуются эритроциты
необратимо серповидной формы,
склонные к агрегации, что повышает
вязкость крови, вызывает закупорку
сосудов (вазоокклюзию), нарушение
микроциркуляции и боль.

В
этом случае точечная мутация обусловливает
нарушение структуры гемоглобина,
гемолиз, анемию, а также закупорку
сосудов и нарушение кровообращения.

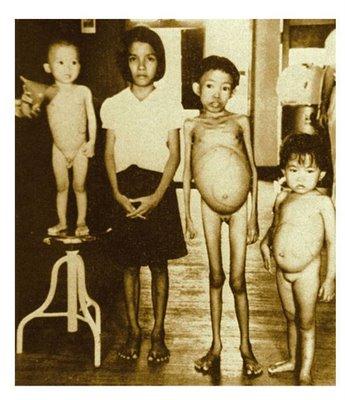
Больные
СКА имеют типичный вид: удлиненный
нижний сегмент тела, выступающий лоб,
«башенный» череп, гепато-спленомегалия.
Самый характерный криз для этого
заболевания — вазоокклюзионный,
проявляющийся резкой болью.Окклюзия
сосудов может развиваться в разных
органах, поэтому клинические симптомы
чрезвычайно разнообразны. В мазках
крови можно обнаружить серповидные
клетки.
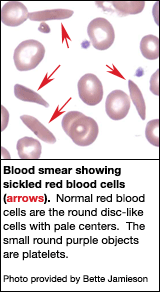
Примерно
одна треть обитателей тропических и
субтропических регионов Африки (т. наз.
«малярийного пояса») являются носителями
признака серповидноклеточности. Они
не страдают от гемолитической анемии,
в то же время более устойчивы по отношению
к малярии.
Талассемии
— замедление или отсутствие синтеза
одной из цепей глобина:
α-талассемия,
ß-талассемия.
При
талассемиях мутации
располагаются не в структурных генах,
а в генах-регуляторах,
поэтому структурных нарушений нет, а
результатом мутаций служит замедление
или отсутствие синтеза одной из глобиновых
цепей и замена ее синтезом другой цепи.
Талассемия встречается в странах
Средиземноморья, в Китае, Индии, в
Европе, у жителей Закавказья и Средней
Азии. Самая высокая заболеваемость –
на Мальдивах, где носители признака
составляют 18%. Гетерозиготная
бета-талассемия наблюдается у 7— 10%
населения
в низменных районах Азербайджана.
Альфа-талассемия
– полное или частичное прекращение
синтеза α-цепей. Компенсаторно
синтезируются: а) в пренатальный период
γ-цепи — образуется тетрамер γ (Hb Барт);
б) в постнатальный – тетрамер ß (HbH).
Синтез
α-цепей кодируют 4 гена, поэтому степень
нарушения их синтеза меньше, чем при
ß-талассемии; выраженный дисбаланс
развивается только тогда, когда поражены
все 4 гена. Агрегаты из ß-цепей более
растворимы, чем агрегаты из α -цепей,
поэтому гемолиз при α-талассемии выражен
слабее, чем при ß-талассемии, а эритропоэз
более эффективен.
Бета-талассемияобусловлена снижением скорости синтеза
ß-цепей гемоглобина (ß+-талассемия)
или отсутствием их синтеза (ß0-талассемия).
Неповрежденные α-цепи избыточно
накапливаются в клетках, что ведет к
повреждению мембраны и разрушению
эритроидных клеток в костном мозге
(неэффективный эритропоэз)и
эритроцитов в крови. Деструкция
эритроидных клеток способствует
гиперплазии костного мозга,
что отражается на структуре скелета,
ведет к повышенному всасыванию железа
и перегрузке организма железом.
Тяжелая
гомозиготная форма ß-талассемии —
болезнь Кули,
или большая талассемия. Кроме того,
выделяют промежуточную, малую и
минимальную талассемию.
 Для
Для
тяжелых форм талассемий характерна:
значительная
спленомегалия,
желтушность,
хронические язвы на нижних конечностях,
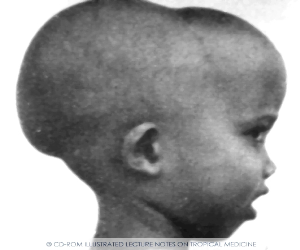
башенный череп,
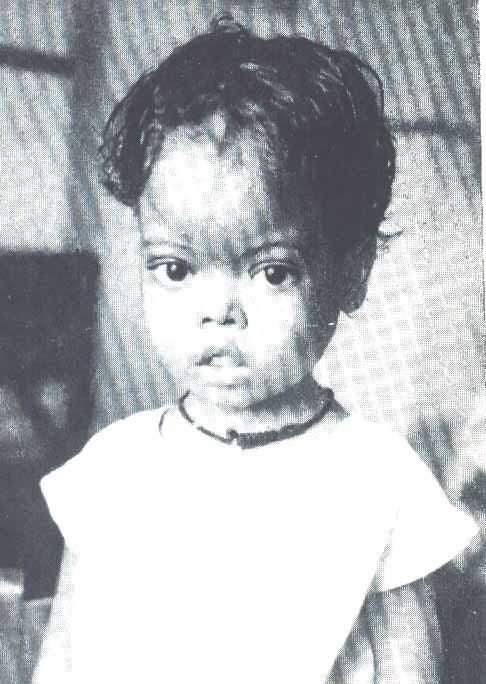
уплощенная
переносица. Скулы выступают, глазные
щели сужены, нарушены прикус и расположение
зубов.
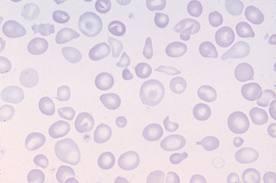
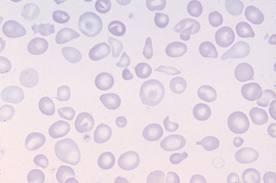
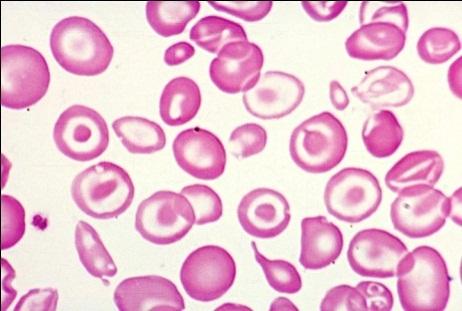 Картина крови: мишеневидные эритроциты,
Картина крови: мишеневидные эритроциты,
анизоцитоз, пойкилоцитоз
.
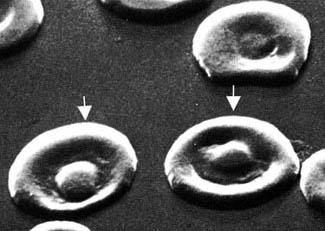
Сканирующая
электронограмма. Стрелками показаны
два кодоцита («хвостатые клетки») – это
другое название мишеневидных клеток.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник

Гемоглобинопатии – это группа тяжелых наследственных заболеваний крови, обусловленных нарушением структуры гемоглобина или снижением синтеза одной и более глобиновых цепей. Клиническая картина крайне разнообразна. Общими симптомами являются гемолитическая анемия, увеличение селезенки, поражение костей. Диагностика осуществляется с помощью микроскопии мазка периферической крови, электрофореза гемоглобина, генетических исследований. Для лечения применяют переливание компонентов крови, препараты гидроксимочевины, инфузионную терапию. У тяжелых пациентов выполняется спленэктомия, аллотрасплантация стволовых клеток.
Общие сведения
Гемоглобинопатии – ряд врожденных гемолитических анемий, характеризующихся изменением аминокислотной последовательности гемоглобина или подавлением образования цепей глобина. Данные патологии нередко заканчиваются летально уже в раннем детском возрасте. Известно около 50 видов гемоглобинопатий. Самыми частыми и опасными для жизни считаются серповидно-клеточная анемия (СКА), талассемии. Гемоглобинопатии распространены на территории Центральной Африки, Южной Азии и наблюдаются преимущественно у лиц негроидной расы. Талассемии также встречаются в странах Средиземноморья. Ежегодно рождается около 350000 детей с дефектами гемоглобина.

Причины гемоглобинопатий
Гемоглобинопатии относятся к аутосомно-рецессивным генетическим заболеваниям. Качественные гемоглобинопатии развиваются вследствие мутаций генов, ответственных за синтез определенных аминокислот в бета-цепи глобина. В результате происходит замена одной аминокислоты на другую (глутаминовой кислоты на валин, лизин и пр.). Это приводит к образованию аномального гемоглобина, гораздо менее растворимого, чем нормальный гемоглобин А, придающего красным кровяным тельцам иную форму (мишеневидную, серповидную), что нарушает их функции и уменьшает продолжительность жизни.
Количественные гемоглобинопатии обусловлены мутацией генов, которые кодируют целую цепь глобина (чаще альфа и бета). При этом сдвигается баланс между глобиновыми цепями – при недостаточном синтезе альфа-цепей возникает избыток бета-цепей, и наоборот. Уменьшаются размеры эритроцитов, в них снижается содержание гемоглобина, а мембрана становится более подверженной различным повреждениям.
Существуют факторы, провоцирующие тяжелые приступы (кризы). К ним относятся обезвоживание, переохлаждения, инфекции, сопровождающиеся высокой лихорадкой. У женщин обострения нередко развиваются на фоне беременности. Но главный патологический стимул качественных гемоглобинопатий – уменьшение концентрации в крови кислорода (гипоксия). Это может произойти, например, при подъеме на большую высоту (восхождение на гору, полет на самолете), где снижено парциальное давление кислорода воздуха, или при тяжелых болезнях дыхательной системы (пневмонии).
Патогенез
Гемоглобинопатии имеют сходные патогенетические механизмы. Измененная структура гемоглобина предрасполагает к интенсивному гемолизу. Длительно текущая анемия способствует компенсаторной гиперплазии костного мозга. Возникает деформация костей черепа, искривление позвоночника. Развиваются экстрамедуллярные очаги кроветворения, приводящие к увеличению размеров печени и селезенки (гепатоспленомегалии). Вследствие спленомегалии наступает гиперспленизм – усиленная деструкция красных клеток крови синусоидами селезенки.
Из-за регулярного гемолиза эритроцитов печень секретирует в желчь большое количество билирубина, что создает условия для образования камней желчного пузыря. У больных гемоглобинопатиями часто возникает перегрузка железом, как за счет постоянных переливаний крови, так и из-за повышения абсорбции железа желудочно-кишечным трактом. Большое количество железа в тканях усиливает процессы перекисного окисления липидов, что повреждает различные органы.
При качественных гемоглобинопатиях под влиянием сниженного содержания кислорода в крови молекулы нерастворимого аномального гемоглобина растягивают мембрану эритроцитов, что приводит к изменению их формы. Деформированные эритроциты хуже переносят кислород, а также способны приклеиваться к сосудистому эндотелию, тем самым закупоривая мелкие сосуды, вызывая тромбозы, окклюзии и инфаркты.
Классификация
Гемоглобинопатии подразделяются на качественные, обусловленные нарушением структуры (последовательности аминокислот) гемоглобина, и количественные, характеризующиеся снижением образования глобиновых цепей. Качественные гемоглобинопатии представлены следующими формами:
- Серповидно-клеточная анемия (Гемоглобинопатия S). Наиболее частый вид. Подразделяется на гомозиготную форму (собственно СКА) с яркой клинической симптоматикой и гетерозиготное носительство (серповидно-клеточную аномалию), имеющее бессимптомное или легкое течение.
- Гемоглобинопатия С. Клиника схожа с СКА, но менее выражена. Отличается большей степенью спленомегалии, чем при СКА.
- Гемоглобинопатия CS (Африканский ревматизм). По течению напоминает СКА. Преобладают приступообразные костно-суставные боли.
- Гемоглобинопатии Е и D. Протекают с небольшой гемолитической анемией.
- Наследственная метгемоглобинемия. При этой разновидности под влиянием различных факторов образуется окисленная форма гемоглобина – метгемоглобин, который более стойко связывается с кислородом и не отдает его тканям.
- Анемия, вызванная носительством нестабильных гемоглобинов. Это доброкачественная патология, при которой возникает незначительная гемолитическая анемия после приема сульфаниламидных препаратов.
К количественным аномалиям гемоглобина относят:
- Бета-талассемии. Наиболее распространенный вариант. Подразделяется на малую талассемию (гетерозиготное носительство с бессимптомным течением или легкой гемолитической анемией) и большую талассемию (анемию Кули) с развернутой тяжелой клинической картиной.
- Альфа-талассемии. Течение может быть различным в зависимости от количества мутантных генов. В основном сходны с гетерозиготной бета-талассемией.
- Синдром водянки плода c гемоглобином Барт (Hb Bart’s). Наиболее тяжелый вид альфа-талассемии. Ребенок погибает внутриутробно.
- Гемоглобинопатию H. Благоприятная малосимптомная форма альфа-талассемии.
- Бета-дельта-талассемии. Практически неотличима от бета-талассемии.
- Гемоглобинопатию Lepore. Эта форма развивается вследствие слияния бета-цепей глобина и схожа с бета-талассемией.
Симптомы гемоглобинопатий
Клиническая картина гемоглобинопатий разнообразна. Гетерозиготные больные имеют либо бессимптомное, либо легкое течение. Гомозиготные формы начинают себя проявлять уже с раннего детства (6 месяцев — 1 год). Общая симптоматика включает признаки гемолитической анемии (бледность, желтушность кожи и слизистых, увеличение селезенки), патологию развития скелета – башенный череп (четырехугольный), уплощенная переносица, искривленный позвоночник.
Из-за повышенной секреции билирубина в желчь могут беспокоить симптомы желчнокаменной болезни уже в детском возрасте – тяжесть или ноющие боли в правом подреберье, приступы желчной колики, обесцвечивание кала. На коже голеней часто возникают длительно незаживающие язвы. Для болезней с дефектом в структуре гемоглобина характерно кризовое течение. Самые тяжелые приступы, нередко заканчивающиеся летально, встречаются при серповидно-клеточной анемии.
Вазоокклюзивный криз
Наиболее типичный. Происходит закупорка мелких сосудов различных органов. Дети испытывают боли в длинных трубчатых костях, у них отекают кисти, стопы, что затрудняет движения лучезапястных, голеностопных суставов (hand-foot синдром). Микротромбозы сосудов кишечника вызывают абдоминальные боли. У молодых мужчин нередко развивается приапизм, впоследствии приводящий к эректильной дисфункции. Кризу сопутствует лихорадка, тахикардия, потливость.
Гемолитический криз
При гемолитическом кризе происходит массивное разрушение красных телец с резким снижением содержания гемоглобина, эритроцитов в крови. Усиливается имеющаяся желтушность, бледность кожных покровов, слизистых, возникают лихорадка, поясничные и абдоминальные боли, присоединяются симптомы снижения артериального давления (головокружение, обмороки). Моча приобретает темный цвет за счет большого количества гемоглобина.
Секвестрационный криз
Во время секвестрационного криза также стремительно падает уровень гемоглобина. Но это происходит не из-за гемолиза, а вследствие венозного тромбоза и скопления большого количества крови в расширенных синусоидах печени и селезенке, что и приводит к обеднению общего кровотока. Кроме симптомов тяжелой анемии по причине выраженной гепатоспленомегалии появляются сильные и распирающие боли в левом и правом подреберье.
Апластический криз
Достаточно редкая форма криза. Она развивается при инфицировании парвовирусом В19, который способен угнетать костномозговое кроветворение. Это сопровождается стремительным (но обратимым) снижением концентрации не только эритроцитов, но и всех других клеток периферической крови (лейкоцитов, тромбоцитов). Поэтому к признакам анемии присоединяется геморрагический синдром (кровотечения из носа, десен), различные инфекции (в основном ОРВИ).
Осложнения
Общими осложнениями гемоглобинопатий считаются ЖКБ, патологические переломы длинных трубчатых костей. Гетерозиготные формы редко сопровождаются неблагоприятными событиями, так как имеют легкое течение. При количественных гемоглобинопатиях из-за отложения избыточного количества железа во внутренних органах развивается сердечная недостаточность, цирроз печени, сахарный диабет 2 типа.
Качественные патологии гемоглобина характеризуются широким спектром неблагоприятных последствий. Наиболее опасными считаются эмболия легочных сосудов, инфаркт миокарда, ОНМК, которые примерно у 10% пациентов приводят к смерти. Закупорка микрососудов, питающих кости, ведет к асептическому некрозу головки бедренной кости (АНГБК). Вследствие постоянных инфарктов селезенки возникает функциональный асплезнизм, из-за чего часто развиваются бактериальные инфекции (бронхиты, пневмонии) с тяжёлым течением, нередко с летальным исходом.
Диагностика
Курацией больных с гемоглобинопатиями занимаются врачи-гематологи, генетики. Во время общего осмотра обращается внимание на цвет кожных покровов (бледность, желтуха), конституциональные нарушения (задержку нервно-психического, физического развития ребенка, аномалии строения скелета). Дополнительное обследование включает:
- Анализы крови. Для общего анализа крови пациентов с качественными гемоглобинопатиями типична нормоцитарная анемия средней степени тяжести, для больных с количественными гемоглобинопатиями – тяжелая микроцитарная анемия. В мазке крови обнаруживаются измененные деформированные эритроциты (серповидные, сферические). В биохимическом анализе крови повышены концентрации свободного железа, ферритина и непрямого билирубина.
- Электрофорез гемоглобина. Основной метод диагностики патологий гемоглобина, широко применяемый в клинической гематологии, который определяет соотношение фракций гемоглобина – электрофорез на пленке с ацетатом целлюлозы или лимоннокислым агаром. Выявляется высокое содержание аномальных гемоглобинов (HbS, HbF, HBA2).
- Генетические исследования. ДНК-тестирование методом ПЦР выполняется только в рамках пренатальной диагностики с целью генетического консультирования семей, имеющих повышенный риск развития патологий гемоглобина. Образцы ДНК получают путем амниоцентеза на 8-10 и 14-16 неделе беременности. Определяют наличие мутаций различных генов.
- Инструментальные исследования. На рентгенографии костей конечностей отмечаются признаки разрастания костного мозга – истончение кортикального слоя, участки остеопороза, расширение костномозгового канала. На рентгене костей черепа выявляется игольчатый периостоз («феномен волосатого черепа»). При УЗИ брюшной полости обнаруживаются гепатоспленомегалия, камни в желчном пузыре.
Гемоглобинопатии дифференцируют с другими врожденными гемолитическими анемиями (мембранопатиями, ферментопатиями, микросфероцитарной анемией Минковского-Шоффара). Постоянные тромбозы нужно дифференцировать от различных тромбофилий. Перегрузку железом следует отличать от наследственного гемохроматоза. Анемия, оссалгии требуют исключения злокачественных миелопролиферативных заболеваний.
Лечение гемоглобинопатий
Людям, страдающим гемоглобинопатиями, требуется проведение сложной многокомпонентной терапии, поэтому все пациенты с гомозиготными формами подлежат обязательной госпитализации в гематологический стационар. Больным с гетерозиготными формами лечение не показано. Основные принципы ведения качественных и количественных патологий гемоглобина несколько отличаются друг от друга.
Консервативная терапия
Подбор консервативной терапии производится с учетом вида гемоглобинопатии, течения заболевания, наличия тех или иных осложнений. Оценивается как клиническая симптоматика, так и лабораторные данные (главным образом, показатели красной крови). Выделяют следующие направления лечения:
- Купирование кризов. При вазоокклюзивных кризах используются обезболивающие препараты (нестероидные противовоспалительные средства), а при выраженном болевом синдроме — наркотические анальгетики. Также для подавления преципитации деформированных эритроцитов назначаются ингаляции кислородом, пероральная или внутривенная регидратация.
- Предупреждение кризов. Для постоянного приема больным качественными гемоглобинопатиями назначается гидроксимочевина. Этот препарат стимулирует образование фетального гемоглобина, подавляющего экспрессию гена, ответственного за синтез аномальных нерастворимых гемоглобинов, что уменьшает склонность эритроцитов к деформации, снижает частоту кризов.
- Терапия осложнений. Инфекционные осложнения до получения результатов бактериологических исследований лечат антибактериальными препаратами, активными против пневмококка, гемофильной палочки, менингококка. Используются антибиотики из группы пенициллинов (амоксициллин). При развитии тромбозов применяются антикоагулянты (низкомолекулярные, нефракционированные гепарины).
- Гемотрансфузии. Так как анемия у пациентов с количественными патологиями гемоглобина всегда тяжелая, основу лечения составляют регулярные гемотрансфузии. Людям, страдающим качественными гемоглобинопатиями, переливания крови проводятся только при секвестрационных, гемолитических, а также апластических кризах.
- Борьба с перегрузкой железа и дефицитом фолатов. Для выведения избытка железа из организма используются хелатирующие средства (дефероксамин). Этот препарат обычно назначается вместе с аскорбиновой кислотой, так как она потенцирует хелатирующее действие дефероксамина. Из-за постоянного гемолиза у больных повышен расход фолатов, поэтому им показан длительный прием больших доз фолиевой кислоты.
Хирургическое лечение
Для ряда пациентов с выраженными признаками гемолиза эффективным лечением является спленэктомия – оперативное удаление селезенки. Другой хирургический вид лечения, позволяющий добиться полной ремиссии – аллотрансплантация гемопоэтических стволовых клеток. Однако этот метод применяется редко, только в очень тяжелых случаях, так как он сопряжен с высокой частотой летальных исходов. При холелитиазе выполняется холецистэктомия.
Экспериментальное лечение
В настоящее время ведутся клинические исследования по поиску лекарства для излечения гемоглобинопатий. Есть успешные результаты генной терапии СКА. Суть лечения заключается во введении в стволовые клетки пациента гена, кодирующего синтез нормальной бета-глобиновой цепи, с помощью обезвреженного лентивируса. Препарат называется LentiGlobin BB305. Его применение привело к улучшению показателей крови, что позволило отказаться от постоянной стандартной терапии. Также проводятся испытания данного препарата при бета-талассемии.
Прогноз и профилактика
Гемоглобинопатии являются тяжелыми заболеваниями с опасными жизнеугрожающими осложнениями. Пациенты с гомозиготной альфа-талассемией умирают еще до рождения в утробе матери. Больные бета-талассемией погибают до наступления пубертатного возраста от сердечной недостаточности. Люди, имеющие качественные гемоглобинопатии, при грамотном лечении могут прожить дольше 50 лет. Основная причина смерти – бактериальные инфекции, тромботические осложнения. При гетерозиготных формах заболевания в большинстве случаев продолжительность жизни не отличается от общей популяции.
Первичная профилактика проводится среди семей, имеющих высокий риск развития гемоглобинопатий. Она заключается в пренатальной диагностике с прерыванием беременности по медпоказаниям. Пациенты, страдающие качественными аномалиями гемоглобина, обязательно должны получить вакцину от гемофильной палочки, пневмококка, менингококка. Детям от 4 месяцев до 6 лет показано длительное применение пенициллиновых антибиотиков для профилактики инфекций. То же касается больных, перенесших спленэктомию.
Источник

