Что такое анемия кули
КУЛИ АНЕМИЯ
(описана американским педиатром Th. В. Cooley, 1871–1945; синонимы – большая бета-талассемия, средиземноморская анемия) – наследственное заболевание из группы гемоглобинопатий с нарушением синтеза нормального гемоглобина и высоким содержанием фетального гемоглобина, обусловливающее дегенеративные изменения и ускоренное разрушение эритроцитов. Характерны: прогрессирующая анемия (проявляется одышкой, сердцебиением); задержка роста; желтуха; гепатоспленомегалия; деформация костей черепа; типичный внешний вид – башенный череп, широко расставленные глаза, монголоидный разрез глаз, плоский нос, большая верхняя челюсть.
Моча имеет бурую окраску и богата уробилином. При обследовании выявляют гипохромную анемию, макроцитоз, ретикулоцитоз, мишеневидные эритроциты. Рентгенологически в теменных и лобных костях выявляют гиперостоз с характерной игольчатой структурой, напоминающий «ежик» или «щетку», в длинных трубчатых костях расширены костномозговые каналы вследствие увеличения объема костного мозга, кортикальный слой истончен; часто наблюдаются патологические переломы. Тип наследования – аутосомно-рецессивный. У гетерозигот может наблюдаться небольшая анемия, не требующая лечения. Лечение гомозигот включает переливания эритроцитарной массы, терапию десмопрессином для профилактики гемосидероза, по показаниям – аллотрансплантацию костного мозга. Прогноз неблагоприятный, заболевание приводит к летальному исходу в детстве или юности. Возможна пренатальная диагностика заболевания.
T. B. Cooley, P. Lee. Series of cases of splenomegaly in children with anemia and peculiar bone changes. Transactions of the American Pediatric Society, 1925; 37: 29.
Энциклопедический словарь по психологии и педагогике.
2013.
Смотреть что такое «КУЛИ АНЕМИЯ» в других словарях:
Кули анемия — (Th. В. Cooley, 1871 1945, амер. педиатр) см. Талассемия большая … Большой медицинский словарь
Кули — Кули: Содержание 1 Топоним 1.1 Белоруссия 1.2 Россия 1.3 Украина … Википедия
анемия — и; ж. [от греч. an не , без и haima кровь]. Мед. Малокровие. ◁ Анемический, ая, ое. А ое состояние. * * * анемия (от греч. an отрицательная приставка и háima кровь) (малокровие), группа заболеваний, характеризующихся уменьшением количества… … Энциклопедический словарь
анемия Кули — β thalassemia, Cooley s anemia β талассемия, анемия Кули. НЗЧ, форма гемолитической анемии <hemolytic anemia>, обусловленное мутациями гена бета гемоглобина (различного типа например, приводящие к образованию гемоглобина Лепора <Lepore… … Молекулярная биология и генетика. Толковый словарь.
Анемия Кули (Cooley ‘S Anaemia) — см. Талассемия. Источник: Медицинский словарь … Медицинские термины
АНЕМИЯ КУЛИ — (Cooley s anaemia) см. Талассемия … Толковый словарь по медицине
Талассемия (Thalassaemid), Анемия Кули (Cooley’S Anaemia) — наследственное заболевание крови, широко распространенное в странах Средиземноморья, Азии и Африки; характеризуется наличием аномалии в белковой части молекулы гемоглобина. В результате пораженные красные клетки крови не могут нормально выполнять … Медицинские термины
ТАЛАССЕМИЯ, АНЕМИЯ КУЛИ — (Cooleys anaemia) наследственное заболевание крови, широко распространенное в странах Средиземноморья, Азии и Африки; характеризуется наличием аномалии в белковой части молекулы гемоглобина. В результате пораженные красные клетки крови не могут… … Толковый словарь по медицине
Талассемия — I Талассемия (thalassaemia; греч. thalassa море + haima кровь) группа наследственных гемолитических анемий, характеризующихся нарушением в различной степени синтеза глобиновых цепей гемоглобина, а также гемолизом, гипохромией и неэффективным… … Медицинская энциклопедия
талассемия большая — (t. major; син.: анемия средиземноморская, анемия эритробластическая, Кули анемия) гомозиготная форма Т., характеризующаяся выраженной гипохромной анемией, наличием мишеневидных эритроцитов, увеличением селезенки, утолщением длинных трубчатых… … Большой медицинский словарь
Источник
Талассемия – это патология крови, для которой характерны нарушения в формировании одной или сразу нескольких цепей гемоглобина в клетках эритроцитов. Это становится причиной развития малых, некачественных эритроцитов.
При таком заболевании наблюдается ухудшение транспортировки кислорода к тканям и клеточным элементам организма. Это вызывает их гипоксию, становятся очевидными проявления признаков болезни.
Раньше недуг чаще наблюдался в климатически жарких местах, где люди страдают малярией: Юго-Восточной Азии, Африке, странах Средиземноморья. Но в современном мире он диагностируется в самых разных регионах из-за миграции населения. Патология формируется одинаково у лиц женского и мужского пола.
.jpg)
Ежегодно рождается около 200 000 детей с этой болезнью, у 50% из них диагностируется гомозиготная форма β-талассемии. Последняя быстро приводит к смерти.
При гетерозиготной форме болезнь себя не проявляет, это возможно только в экстремальных ситуациях. Однако высок риск ее развития у потомков, они могут получить ее как наследственную патологию.
Причины талассемии
Они изучены хорошо, поскольку заболевание достаточно распространено. Достоверно известно, что первые изменения наблюдаются при нарушении синтеза белковых цепей гемоглобина, который способен связываться с молекулами кислорода и транспортировать их ко всем тканям организма.
Чаще всего свое развитие заболевание получает из-за генов, которые дети наследуют от своих родителей. В тех из них, которые несут ответственность за синтез гемоглобина, обнаруживается мутация.
При изучении структуры обнаруживается пигмент, в состав которого входит железо и 2 пары соединений белка: альфа- и бета-цепи. Если синтез одной из них нарушается, то накапливается вторая.

Провоцируют такие изменения те гены, что несут ответственность за формирование белка гемоглобина. При такой мутации идет нарушение правильного набора аминокислот в цепочке. Явление характерно и для возбудителя малярии.
Наследственность у ребенка способна проявиться в двух вариантах:
- Гомозиготный вид патологии – от мамы и папы одновременно.
- Гетерозиготный – от одного из родителей.
Классификация
Если аномальный белок гемоглобина наследуется с обеих родительских сторон, то это тяжелая форма, для которой характерны ярко выраженные проявления. Это большая талассемия, или болезнь Кули (в честь доктора, что ее описал).
.jpg)
Она наследуется по типу рецессивному (двухаллельная система), в основе лежит снижение синтеза цепей полипептидов.Если это идет со стороны одного родителя, то при отсутствии крайне неблагоприятных обстоятельств признаки отсутствуют.
В современной классификации выделены следующие формы недуга:
- Альфа-талассемия. Наблюдается при отсутствии одного либо нескольких генов, которых в норме должно быть по два от матери и отца. Именно такое количество обеспечивает достаточную выработку белков цепочки альфа-глобина. При отсутствии двух генов возможно проявление симптомов анемии. При недостатке трех генов диагностируется гемоглобинопатия H, которая ярко выраженно проявляется симптомами малокровия. Отсутствие четырех генов – явление довольно редкое, которое носит название “водянка плода”. Такие дети умирают до, во время либо сразу после родов.
- Бета-талассемия наблюдается при нарушении одного либо двух генов, которых в норме идет по одному от папы и мамы. Вырабатывается недостаточное количество белка бета-глобина. При видоизменении одного гена человек является носителем болезни, не исключено проявление умеренного малокровия. При изменении обоих генов одновременно диагностируется бета-талассемия интермедия (анемия Кули). При основной развивается и тяжелая форма анемического синдрома. Гомозиготная бета-форма выявляет себя к году жизни ребенка и различается проявлением признаков. Гетерозиготная (малая форма талассемии) характеризуется размытыми симптомами, либо признаки полностью отсутствуют.
Нужно учитывать, что в отдельных случаях гомозиготная форма протекает легко, ее нельзя квалифицировать как большую талассемию, поэтому ее отмечают как промежуточную.
У этой патологии есть три степени:
- Тяжелая. Аномальные изменения настолько сильны, что ребенок умирает на первом году жизни.
- Среднетяжелая. Ребенок способен прожить около восьми лет.
- Легкая форма. Взрослый человек умирает, не дожив до старости.
Несмотря на общий диагноз, β-талассемия у людей может значительно различаться результатами исследований, проявлениями признаков, клиническим течением, лечением и прогнозами на будущее.
Симптомы и проявления
Признаки зависят от формы и времени появления мутации. Большая талассемия случается при гомозиготном происхождении, проявляется у детей сразу после рождения:
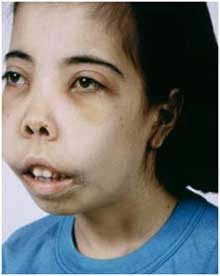
- Удлиненная кверху черепная коробка.
- Верхняя челюсть больше развита по сравнению с нижней.
- Лицевой скелет черепа напоминает монголоидный.
К году жизни наблюдаются следующие признаки:
- Расширение перегородки носа, приплюснутая его форма.
- Формирование аномальных костных наростов в области стоп.
- Нарушения прикуса.
- Желтушность кожных покровов из-за нарушения функции селезенки.
Дефицит кислорода в тканях вызывает такие симптомы:
- Увеличение печени, развитие раннего цирроза.
- Формирование сердечной недостаточности. Излишек железа скапливается на мышце миокарда и нарушает сократительную деятельность мышцы сердца.
- Запаздывание в физиологическом и интеллектуальном развитии.
- Формирование сахарного диабета.
У взрослых людей проявляются следующие симптомы:
- Формирование язв на кожных покровах, их провоцируют аномальные нарушения в кровообращении.
- Частые пневмонии.
- Регулярные переломы костей.
- Воспалительные процессы в желчном пузыре, связанные с отложением камней.
- Кардиосклероз — патология сердца, характеризующаяся разрастанием соединительного рубца в миокарде, замещением мышц и деформацией клапанов.
- Формирование сепсиса при инфекционных патологиях.
- Нарушения на половом уровне.
При развитии промежуточной формы заболевания не наблюдается изменений внешнего вида человека, умственное и физическое развитие в норме, однако есть увеличение селезенки и ломкость костей.
Признаки большой талассемии
Наблюдается сразу после появления на свет:
- Удлиненная форма черепа.
- Монголоидный тип лица.
- Увеличение верхней челюсти.
- Увеличение перегородки носа.
Проведение анализа крови указывает на гепатомегалию, которая заканчивается формированием цирроза и диабета.
.jpg)
Если при рождении ставится диагноз “талассемия”, причем признаки яркие, то чаще всего такие дети могут прожить около года.
Симптомы малой талассемии
Если генная мутация передалась от одного из родителей, то развивается малая талассемия. Симптомы в таком случае размытые либо не проявляются вообще. Клиническая картина следующая:
- Повышенная утомляемость.
- Ухудшение работоспособности.
- Регулярные боли в голове, беспричинные головокружения.
- Бледность кожи с проявлением желтушности.
- Увеличение селезенки.
Наибольшая опасность такого состояния заключается в повышении риска попадания инфекции в организм.
Диагностика заболевания
Обследование включает в себя несколько этапов:
- Сбор анамнеза. Определяются опасные патологии, которые диагностировались у женщины при беременности и могли стать причиной развития патологии.
- Оценка протекания периода вынашивания ребенка.
- Тщательный осмотр врачом: оценивается общее состояние пациента, состояние и цвет кожи, проводится пальпация брюшной области для определения гепатоспленомегалии.
- Опрос родителей либо самого пациента для выяснения выраженности симптомов и тяжести заболевания.
Также назначается проведение следующих лабораторных исследований:
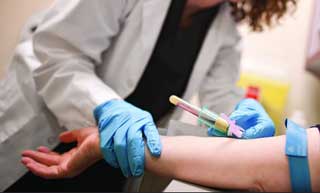
- Биохимический анализ крови.
- ПЦР-тесты.
- Исследование биологического синтеза цепочек гемоглобина.
- Общее исследование крови.
- Электрофорез гемоглобина.
Также необходимо проведение инструментальных методов диагностики:
- УЗИ брюшной полости (обнаруживает увеличение селезенки).
- Рентген костей.
- Пункция костного мозга.
- Развитие плода в утробе матери.
При обнаружении гомозиготной формы патологии назначается искусственное прерывание беременности.
Лечение
Определив, что это такое и каковы признаки, необходимо выбрать методы терапии. Лечебные меры в первую очередь направляют на нормализацию количества гемоглобина.
Помимо этого, есть и другие методы лечения:
.jpg)
- При тяжелых формах недуга необходимо проводить частое переливание крови, но это дает временный результат.
- Назначается хелат железа.
- В современной медицине есть возможность проводить переливание размороженных либо отфильтрованных эритроцитов. Такая процедура более безопасна для детей.
- При увеличенной селезенке назначается ее удаление. Хирургическое лечение возможно только после 5 лет жизни, необходимо учитывать возрастание риска проникновения инфекции.
- Трансплантация костного мозга, но найти донора проблематично.
К клиническим рекомендациям относится специальная диета, которая подразумевает прием орехов, чая, сои, какао (продуктов, которые противостоят усвоению железа). Прием аскорбиновой кислоты, которая способствует выводу железа из организма.
Возможные осложнения
При умеренных и тяжелых формах лечебные меры направлены на продление периода жизни и устранение возможных осложнений, таких как:
- Патологии сердца и печени. Высокий уровень железа в организме может стать причиной повреждения различных тканей, больше всего этому подвержены сердечная мышца и печень. Это нередко становится причиной смертельного исхода, поэтому необходимо проведение регулярного переливания крови.
- Проникновение инфекции в организм также нередко является причиной смерти пациента, особенно это касается тех, у кого проводилась операция по удалению селезенки.
- Остеопороз. При такой патологии костные элементы подвержены частым переломам.
Профилактика
Вылечить талассемию невозможно, поэтому необходимо принимать меры профилактики, которые заключаются в следующем:
.jpg)
- Осуществление диагностики во время беременности, особенно если у обоих родителей наблюдается талассемия.
- Проведение генетического исследования.
- При необходимо проводится искусственное прерывание беременности.
Прогнозы
При диагностировании малой талассемии прогнозы хорошие, человек ведет нормальную жизнь, продолжительность которой практически как у здорового.
При бета-талассемии лишь некоторые больные способны дожить до половозрелого возраста.
Тяжелая гомозиготная форма требует постоянного лечения, поскольку только такие мероприятия, как переливание крови, поддерживают жизнь пациента.
Мы настоятельно рекомендуем не заниматься самолечением, лучше обратитесь к своему лечащему доктору. Все материалы на сайте носят ознакомительный характер!
Источник
Талассеми́я (анемия Кули) — заболевание, наследуемое по рецессивному типу (двухаллельная система), в основе которого лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина. В норме основным вариантом (97 %) гемоглобина взрослого человека является гемоглобин А. Это тетрамер, состоящий из двух мономеров α-цепей и двух мономеров β-цепей. 3 % гемоглобина взрослых представлено гемоглобином А2, состоящим из двух альфа- и двух дельта-цепей.
Существуют два гена HBA1 и HBA2, кодирующих мономер альфа, и один HBB-ген, кодирующий мономер бета.
Наличие мутации в генах гемоглобина может привести к нарушению синтеза цепей определённого вида.
Классификация[править | править код]
В зависимости от того, синтез какого из мономеров нарушен, разделяют альфа-, бета- и дельта-талассемию.
По тяжести клинических проявлений выделяют тяжёлую, среднюю и лёгкую формы заболевания.
Альфа-талассемия[править | править код]
Связана с мутациями в генах HBA1 и HBA2. Есть всего 4 локуса, кодирующего α-цепи. Наличие мутации в одном из локусов приводит к минимальным клиническим проявлениям. Нарушения в двух локусах выражаются лёгкой формой анемии. При мутациях в трёх локусах возникает значительное уменьшение продукции α-глобина. При этом избыточные цепи β-глобина образуют тетрамеры — гемоглобин Н. Эта форма носит также название гемоглобинопатии Н. Характер заболевания может варьироваться от лёгкой до тяжёлой картины гипохромной микроцитарной анемии. Присутствие мутаций во всех четырёх аллелях альфа-глобина не совместимо с жизнью. Ребёнок с такой патологией погибает внутриутробно или вскоре после рождения. Из пуповинной крови таких детей можно выделить гемоглобин Барта.
Бета-талассемия[править | править код]
Существует два варианта бета-талассемии — большая талассемия CD8(-AA) и малая талассемия (minor), из которых большая талассемия — наиболее тяжёлая форма заболевания. Возникает при наличии мутаций в обоих аллелях гена бета-глобина. В отсутствие или при резком уменьшении производства бета-цепей гемоглобин А вытесняется гемоглобином F, в норме вырабатывающимся у плода и сменяющимся на гемоглобин А после родов.
Малая талассемия связана с наличием мутации в одном из аллелей гена бета-глобина. Как правило, протекает легко и не требует лечения.
Этиология[править | править код]
Талассемию вызывают точечные мутации или делеции в генах гемоглобина, ведущие к нарушению синтеза РНК, что приводит к уменьшению или полному прекращению синтеза одного из видов полипептидных цепей. Синтез цепей другого вида продолжается. Это приводит к образованию нестабильных полипептидных агрегатов из избыточных цепей, нарушающих нормальное функционирование эритроцитов, и их разрушению. Повышенный гемолиз эритроцитов вызывает анемию.
Эпидемиология[править | править код]
Альфа-талассемия распространена в Западной Африке и Южной Азии. Бета-талассемия часто встречается в странах Средиземноморья, Западной Азии и Северной Африки. Это регионы, где распространена малярия. Гетерозиготные носители мутаций в генах альфа- и бета цепей гемоглобина являются более устойчивыми к малярийному плазмодию. Имеются очаги талассемии в Азербайджане, в равнинных районах которого гетерозиготная бета-талассемия наблюдается у 7—10 % населения.
Клиническая картина[править | править код]
При талассемии характерны гипохромная анемия, анизоцитоз эритроцитов, наличие мишеневидных форм эритроцитов (пятно гемоглобина в центре клетки, напоминающее мишень). При этом содержание сывороточного железа нормальное или повышенное. Компенсаторная гиперплазия костного мозга ведёт к нарушениям в строении лицевого черепа. Череп может стать квадратным, башенным; нос приобретает седловидную форму; нарушается прикус и расположение зубов. Отмечается желтушность кожи и слизистых оболочек. Селезёнка и печень увеличены. Больные подвержены инфекционным заболеваниям. Рано начавшаяся анемия обуславливает физическое и умственное недоразвитие ребёнка.
Примечания[править | править код]
- ↑ 1 2 Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ 1 2 3 Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
Ссылки[править | править код]
- Талассемия
- Фонд анемии Кули
- «Зинтегло»: генная терапия бета-талассемии
Источник

