Анемия фанкони код мкб
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Профилактика
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Апластическая анемия.
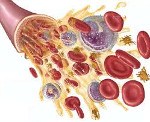
Апластическая анемия
Описание
Апластическая анемия. Угнетение функции кроветворения красного костного мозга (эритроцитопоэза, лейкопоэза и тромбоцитопоэза), приводящее к пангемоцитопении. К основным клиническим симптомам апластической анемии принадлежат головокружение, слабость, обмороки, одышка, покалывание в груди, кожные геморрагии, кровотечения, склонность к развитию инфекционно-воспалительных и гнойных процессов. Апластическая анемия диагностируется на основании характерных изменений гемограммы, миелограммы и гистологического исследования трепанобиоптата. Лечение апластической анемии включает проведение гемотрансфузий, иммуносупрессивной терапии, миелотрансплантации.
Дополнительные факты
Апластическая (гипопластическая) анемия – тяжелое расстройство гемопоэза (чаще всех его звеньев), сопровождающееся развитием анемического, геморрагического синдромов и инфекционных осложнений. Апластическая анемия развивается в среднем у 2 человек на 1 млн. Населения в год. Приблизительно с одинаковой частотой патология поражает мужчин и женщин. Возрастные пики заболеваемости приходятся на возраст 10–25 и старше 50 лет. При данной патологии в костном мозге чаще нарушается образование всех трех типов клеточных элементов крови (эритроцитов, лейкоцитов и тромбоцитов), иногда — только одних эритроцитов; в зависимости от этого различают истинную и парциальную апластическую анемию. В гематологии апластическая анемия относится к числу потенциально фатальных заболеваний, приводящих к гибели 2/3 заболевших.
Апластическая анемия
Причины
По происхождению апластическая анемия может быть врожденной (связанной с хромосомными аберрациями) и приобретенной (развившейся в течение жизни). Принято считать, что угнетение миелопоэза при апластической анемии связано с появлением в красном костном мозге и крови цитотоксических T-лимфоцитов, производящих фактор некроза опухолей и γ-интерферон, которые в свою очередь подавляют ростки кроветворения. Запускать этот механизм могут различные внешнесредовые (химические соединения, физические явления, лекарственные вещества), а также эндогенные факторы (вирусы, аутоиммунные реакции).
Достоверно установлена связь апластической анемии с приемом некоторых противоопухолевых, противосудорожных, антибактериальных, антитиреоидных, противомалярийных препаратов, транквилизаторов, препаратов золота и тд , обладающих потенциальным миелотоксическим эффектом. Лекарственные вещества могут вызывать как прямое повреждение стволовых кроветворных клеток, так и опосредованное — через аутоиммунные реакции. Апластические анемии, связанные с таким механизмом развития, называются лекарственными.
Из вирусных агентов наибольшее значение уделяется возбудителям гепатитов В, С и D. В этом случае апластическая анемия обычно развивается в течение полугода после перенесенного вирусного гепатита. При изучении патогенеза было замечено, что репликация вируса происходит в мононуклеарах крови и костного мозга, а также в иммунных клетках. Предполагается, что подавление миелопоэза в этом случае является своеобразным иммунным ответом, возникающим против клеток, несущих на своей поверхности вирусные антигены. Такой вид апластической анемии выделяется в отдельную форму – постгепатитную. Среди других вирусных инфекций называются ЦМВ, инфекционный мононуклеоз, грипп.
Также описаны случаи апластической анемии, вызванные инфицированием туберкулезом, интоксикацией, лучевой болезнью, лимфопролиферативными заболеваниями (тимомой, лимфомой, хроническим лимфобластным лейкозом), беременностью. Почти в половине наблюдений причину апластической анемии выявить не удается — такие случаи относят к идиопатической форме.
Т. О. , в основе апластической анемии может лежать либо первичное повреждение гемопоэтических стволовых клеток, либо нарушение их эффективной дифференцировки. Основные клинические проявления апластической анемии обусловлены пангемоцитопенией – снижением в составе крови всех ее форменных элементов (эритроцитов, лейкоцитов, тромбоцитов).
Классификация
Кроме различных этиологических вариантов апластической анемии (лекарственного, постгепатитного, идиопатического), различают острую (до 1 мес. Течения), подострую (от 1 до 6 мес. ) и хроническую (более 6 мес. ) форму заболевания. На основании выраженности тромбо- и гранулоцитопении апластическая анемия подразделяется на 3 степени тяжести:
• очень тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,2х109/л).
• тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,5х109/л), по данным трепанобиопсии – низкая клеточность костного мозга (менее 30% от нормы).
• умеренную (тромбоцитов более 20,0х109/л; гранулоцитов более 0,5х109/л).
Апластическую анемию, протекающую с избирательным угнетением эритропоэза, называют парциальной красноклеточной аплазией.
Симптомы
Поражение трех гемопоэтических ростков (эритро-, тромбоцито- и лейкопоэза) обусловливает развитие анемического и геморрагического синдромов, инфекционных осложнений. Дебют апластической анемии обычно происходит остро. Анемический синдром сопровождается общей слабостью и утомляемостью, бледностью кожи и видимых слизистых, шумом в ушах, головокружением, покалыванием в груди, одышкой при нагрузке.
Основным проявлением тромбоцитопении выступает геморрагический синдром. Больные отмечают появление петехий и экхимозов на коже, повышенную кровоточивость десен, спонтанные носовые кровотечения, меноррагии. Возможно возникновение гематурии, маточных и желудочно-кишечных кровотечений. Следствием лейкопении и агранулоцитоза служит частое развитие инфекционных процессов – стоматитов, пневмоний, инфекций кожи и мочевыводящих путей. Для апластической анемий нехарактерны похудание, лимфаденопатия, гепато- и спленомегалия – при этих признаках следует искать другую причину пангемоцитопении. Летальный исход при апластической анемии может быть обусловлен кровоизлияниями во внутренние органы, фатальными кровотечениями, инфекционными осложнениями, анемической комой.
Врожденная апластическая анемия (синдром Фанкони) обычно развивается у детей в возрасте до 10 лет и кроме аплазии костного мозга характеризуется другими нарушениями: микроцефалией, гипоплазией почек, низкорослостью, аномалиями развития верхних конечностей (гипоплазией первой пястной и лучевой кости), гипоспадией, гиперпигментацией кожи, крайней степенью тугоухости и тд.
Диагностика
Гематологическое обследование включает внимательный клинический осмотр и проведение специальных диагностических исследований: общего и биохимического анализа крови, стернальной пункции, трепанобиопсии. При физикальном обследовании выявляется выраженная бледность или желтушность кожи, артериальная гипотония, тахикардия.
Для гемограммы при апластической анемии типичны эритро-, лейкоцито- и тромбоцитопения, нейтропения и относительный лимфоцитоз. Исследование пунктата костного мозга показывает уменьшение количества миелокариоцитов и мегакариоцитов, снижение клеточности; в трепанобиоптате обнаруживается замещение красного костного мозга жировым (желтым). В рамках диагностического поиска апластическую анемию необходимо дифференцировать с мегабластными (В12-дефицитными, фолиеводефицитными) анемиями, идиопатической тромбоцитопенической пурпурой, пароксизмальной ночной гемоглобинурией, острым лейкозом.
Лечение
Больные с апластической анемией госпитализируются в специализированные отделения. Им обеспечиваются полная изоляция и асептические условия для предупреждения возможных инфекционных осложнений. Проведение эффективного лечения апластической анемии является сложной проблемой практической гематологии. Наиболее благоприятные прогнозы на долгосрочную выживаемость оказывает проведение аллогенной трансплантации костного мозга. Однако ввиду сложности подбора иммунологически совместимого донора процедура используется ограниченно. В качестве экспериментальных подходов рассматриваются аутологичные трансплантации, пересадка стволовых клеток периферической крови.
Гораздо чаще при апластической анемии назначается иммуносупрессивная терапия, включающая комбинацию антитимоцитарного иммуноглобулина и циклоспорина А. В комплексе с курсом иммуносупрессивной терапии показано проведение заместительной гемотрансфузионной терапии (переливание тромбоцитов и эритроцитарной массы), плазмафереза. Больным с нетяжелой формой апластической анемии может быть показано проведение спленэктомии, эндоваскулярной окклюзии селезеночной артерии.
Прогноз
Прогноз апластической анемии определяется этиологической формой, тяжестью и остротой течения. Критериями неблагоприятного исхода служат быстрое прогрессирование заболевания, тяжелый геморрагический синдром и инфекционные осложнения. После трансплантации костного мозга ремиссии удается достичь у 75–90% пациентов.
Профилактика
Первичная профилактика апластической анемии предполагает исключение влияния неблагоприятных внешнесредовых факторов, необоснованного применения лекарственных препаратов, предупреждение инфекционной заболеваемости и тд Пациентам с уже развившимся заболеванием требуется диспансерное наблюдение гематолога, систематическое обследование и длительная поддерживающая терапия.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Рубрика МКБ-10: D61.0
МКБ-10 / D50-D89 КЛАСС III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм / D60-D64 Апластические и другие анемии / D61 Другие апластические анемии
Определение и общие сведения[править]
Анемия Фанкони
Синонимы: панцитопения Фанкони
Анемия Фанкони — является наследственным нарушением репарации ДНК, характеризуется прогрессирующей панцитопенией с недостаточностью костного мозга, разнообразными врожденными пороками развития и предрасположенность к развитию гематологических или солидных опухолей.
Последние определение несущей частоты дали оценку более чем 1/200, с ожидаемой распространенности при рождении, по крайней мере, 1 / 160,000. В некоторых популяциях, несущая частота значительно выше, из-за мутации-основателей. Более 2000 случаев заболевания сообщались в литературе.
Анемия Фанкони, как правило, аутосомно-рецессивное заболевание, но может произойти и Х-сцепленная передача.
Этиология и патогенез[править]
Анемия Фанкони происходит из-за мутаций в генах, участвующих в репарации ДНК и геномной стабильности. Пятнадцать генов, представляющих 15 комплементационных групп были идентифицированы на сегодняшний день.
Клинические проявления[править]
У 2/3 больных, первые признаки анемии Фанкони — врожденные пороки развития, которые могут затрагивать скелет, кожу, мочеполовую и сердечно-легочную сферу, желудочно-кишечный тракт и центральную нервную систему. Аномалии конечностей могут быть односторонними или двусторонними, причем в последнем случае часто асимметричными. Также могут присутствовать низкий рост и вес, микроцефалия и/или микрофтальм. Нередки пигментация кожи и гипоплазия основания большого пальца (возвышения тенара). Почти 20% пациентов имеют пороки развития уха с или без потери слуха. Врожденные пороки развития могут варьироваться в семье. Если врожденные пороки развития не манифестированы, постановка диагноза может быть отложена до наступления отказа костного мозга (BMF), которое происходит в среднем возрасте до 7 лет. Гематологические нарушения могут возникать в более молодом возрасте, реже у взрослых, при этом 90% пациентов отказ костного мозга развивается в возрасте до 40 лет. У пациентов может развиться острый миелоидный лейкоз, которому часто предшествует миелодиспластический синдром. Пациенты также высоко предрасположены к образованию солидных опухолей головы, шеи и аногенитальной области. Маленький рост часто обусловлен гормональными дефектами. Фертильность полность нарушена у мужчин и сильно нарушается у половине женщин. Беременность часто осложняется.
Конституциональная апластическая анемия: Диагностика[править]
Анемию Фанкони можно заподозрить при панцитопении и характерных фенотипических проявлениях. Прогрессирующая апластическая анемия обычно развивается в возрасте старше 1 года, в среднем — в возрасте 7 лет. Макроцитоз наблюдается еще до возникновения цитопении. При подозрении на анемию Фанкони необходим поиск сопутствующих аномалий развития. Диагноз подтверждают усиленным разрушением хромосом под действием диэпоксибутана, митомицина С или других веществ, повреждающих ДНК. Возможна пренатальная диагностика.
Дифференциальный диагноз[править]
Клинические проявления анемии Фальконе совпадают со многими нарушениями мальформации (Дубовица, Секеля, Холта-Орама, Баллера-Герольда, тромбоцитопения с отсутствуем радиальной кости, синдром неймегеновского повреждения, VACTERL ассоциация, врожденный дискератоз.
Диагностика анемии Фальконе более очевидна когда у пациента развивается отказ костного мозга или злокачественные опухоли. Анемию Фальконе следует рассматривать в дифференциальной диагностике всех молодых пациентов с отказом костного мозга неизвестной этиологии.
Следует также рассмотреть другие синдромы с предрасположенностью к новообразованиям (синдром Блума, Ротмунда-Томсона или синдром Вернера) или синдромы, сопровождающиеся панцитопенией (анемия Блекфана-Даймонда, иммунная панцитопения, синдром Пирсона или синдром Швахмана-Даймонда).
Конституциональная апластическая анемия: Лечение[править]
Поддерживающая терапия включает в себя переливание эритроцитов или очищенных тромбоцитов. Единственным средством для лечения гематологических проявлений является пересадка гемопоэтических стволовых клеток. Тем не менее, этот подход несет тенденцию к увеличению риска солидных опухолей. Симптоматическое лечение включает в себя пероральное введение андрогенов, что улучшает показатели крови у некоторых пациентов, в частности, кол-во эритроцитов. Назначение факторов роста кроветворения может быть рассмотрено после проведения аспирации и биопсии костного мозга, которые должны выполняться регулярно во время лечения.
Прогноз
Отказ костного мозга и злокачественные опухоли ухудшают прогноз, сокращая продолжительность жизни.
Профилактика[править]
Прочее[править]
Анемия Блекфена-Даймонда
Синонимы: синдром Аазе, врожденная аплазия эритроцитов, врожденная эритроцитарная аплазия, врожденная гипоплатическая анемия типа Блекфена-Даймонда
Определение и общие сведения
Анемия Блекфена-Даймонда является врожденной арегенеративной и часто макроцитарной анемией с эритробластопенией.
Ежегодная заболеваемость в Европе по оценкам составляет около 1 / 150,000. Оба пола в равной степени подвержены заболеванию и ни в одной этнической группе не было выявлено предрасположенности к врожденной гипопластической анемии.
Анемия Блекфена-Даймонда наследуется как аутосомно-доминантный признак с переменной пенетрацией.
Этиология и патогенез
В настоящее время, вызывающие болезнь мутации выявляются у 40-45% пациентов. Все вовлеченные в патогенез анемии Блекфена-Даймонда гены кодируют белки рибосом, среди них есть как небольшие субъединицы (RPS7, RPS17, RPS19, RPS24 ), так и крупные (RPL5, RPL11, RPL35a). Мутации в RPS19, RPL5 и RPL11 наблюдаются у 25%, 9% и 6,5% пациентов соответственно, в то время как другие гены, отвественны за 1-3% случаев. Единственная очевидная корреляция генотип/фенотип — частые краниофациальные аномалии у носителей мутации RPL5 и RPL11 и наоборот эти аномалии редки у носителей мутации RPS19.
Клинические проявления
Анемия Блекфена-Даймонда обнаруживается в раннем возрасте, как правило, в течение первых 2-х лет жизни, возникновение анемии после 4-х лет очень маловероятно. Первые признаки заболевания — бледность и одышка, особенно во время кормления или во время сосания. Бледность, без органомегалии — признака, указывающего на гемолиз или вовлечением других гемопоэтических клеточных линий. Более половины всех пациентов демонстрируют низкий рост и врожденные аномалии, наиболее часто черепно-лицевые (синдром Пьера-Робина и расщепление нёба), большого пальца и урогенитального тракта. Беременность у женщин-носителей признака несет высокий риск, как для матери, так и для ребенка. Пациенты с анемией Блекфена-Даймонда также имеют более высокий риск лейкемии и новообразований.
Диагностика
У ребенка с анемией и эритробластопенией диагноз может быть поддержан семейной историей (10-20% случаев), сопутствующими пороками развития (40% случаев), а также повышенным уровнем эритроцитарной аденозиндезаминазой, который является частым, но неспецифическим признаком, поскольку также может быть повышена у родственников при отсутствии других симптомов заболевания. Обнаружение болезнетворной мутации имеет диагностическое значение.
Генетическое консультирование и пренатальная диагностика затруднена из-за изменчивости клинических проявлений и того факт, что только 40-45% пациентов имеют мутации в пределах гена RP. В семейных случаях, риск рецидива составляет 50%. Проводить ультразвуковое наблюдение во время беременности рекомендуется во всех случаях.
Дифференциальный диагноз
Дифференциальный диагноз должен включать в себя транзиторную эритробластопению, хронические инфекции парвовируса В19 и другие врожденные анемии.
Лечение
Регулярные переливания крови и долгосрочная терапия кортикостероидами. Лечение должно быть адаптировано к каждому случаю и в зависимости от возраста пациента. Стероиды не следует вводить в течение первого года жизни. Низкий рост, как в рамках синдрома так и из-за связанных с лечением осложнений (стероиды, гемохроматоз), является серьезной проблемой для таких пациентов. Аллогенная трансплантация костного мозга должны быть рассмотрена при резистентности к кортикостероидам, если доступен здоровый и HLA-идентичный сиб.
Прогноз
Прогноз, как правило, хороший. Тем не менее, осложнения лечения и более высокий уровень заболеваемости раком может привести к снижению продолжительности жизни. Тяжесть заболевания зависит от качества и реакции на лечение. Для пациентов, проходящих регулярные переливания, качество жизни явно не страдает.
Синдром Швахмана-Даймонда
Синонимы: недостаточность поджелудочной железы и дисфункция костного мозга, синдром Швахмана, синдром Швахмана-Бодиана-Даймонда
Определение и общие сведения
Синдром Швахмана-Даймонда является редким мультисистемным синдромом, который характеризуется хронической и обычно легкой нейтропенией, экзокринной недостаточностью поджелудочной железы со стеатореей, скелетной дисплазией с низким ростом и повышенным риском развития аплазии костного мозга или лейкозной трансформации.
Во всем мире распространенность оценивается примерно 1/350000 и распространенности при рождении на уровне около 1 / 200.000 живорожденных. Передача аутосомно-рецессивная.
Этиология и патогенез
Синдром Швахмана-Даймонда в 95% случаев вызывается мутаций гена SBDS (7q11.22), кодирующим белок рибосом, участвующий в биогенезе рибосом и других клеточных процессах.
Клинические проявления
Синдром Швахмана-Даймонда имеет переменную клиническую картину, даже внутри одной семьи. Как правило, он проявляется в младенчестве или раннем детстве. Наиболее распространенный симптом — интерметтирующая умеренная нейтропения, сопровождаемая развитием рецидивирующих инфекций. Умеренная анемия и тромбоцитопения также могут наблюдаться у пациентов. Экзокринная недостаточность панкреатической железы приводит к снижению прибавки массы тела, замедлении роста и хронической стеаторее. Участие со строны костной ткани характеризуется замедленным созреванием костей, дисплазией метафизов, впалой грудной клеткой и генерализованной остеопенией. Другие симптомы включают в себя экзему или ихтиоз кожи, аномалии зубнов и психомоторную заторможенность. Умеренный или тяжелый интеллектуальный дефицит (50% пациентов) вызывает трудности в обучении. Гематологические проявления могут быть осложнены аплазией костного мозга, острым миелолейкозом и миелодиспластическим синдромом. В неонатальном периоде патология обычно не наблюдается, но в некоторых случаях сообщалось о панцитопении, респираторным дистрессе и тяжелой спондилометафизарной дисплазии.
Диагностика
Диагноз основывается на клинических, лабораторных и рентгенологических исследованиях. Анализ крови демонстрирует наличие нейтропении (абсолютное количество нейтрофилов <1500 / мл), которая может сопровождаться от легкой до умеренной тромбоцитопенией, умеренной анемией и повышением фетального гемоглобина. Экзокринная недостаточность поджелудочной железы проявляется низкими уровнями поджелудочной изоамилазы и/или трипсиногена, низким уровнем фекальной эластазы в анализе кала, жировой дистрофией на МРТ (МРТ картина может быть нормальной до возраста 5 лет). Рентгенография позволяет обнаружить, как правило в возрасте после 5 лет, — аномалии метафизов и пластин роста. Мазок костного мозга выявляют различную степень с дисгранулопоэза или дисэритропоэза. Диагноз подтверждается с помощью генетического тестирования.
Дифференциальный диагноз
Дифференциальный диагноз включает муковисцидоз, синдром Пирсона, анемию Фанкони, синдром Йохансона-Близзарда, анемию Блекфана-Даймонда, целиакию и аутосомно-рецессивную тяжелую врожденную нейтропению вследствие дефицита G6PC3.
Лечение
Недостаточность поджелудочной железы требует назначения панкреатических ферментов и адаптированной диеты. Антибиотикопрофилактика терапия может быть достаточной, чтобы избежать инфекций; в противном случае, может быть предложен гранулоцитов колониестимулирующий фактор. Тяжелые гематологические осложнения требуют трансплантации гемопоэтических стволовых клеток. Хирургическое вмешательство может быть предложено для скелетных аномалий. Пациенты с трудностями в обучении требуют специальной образовательной поддержки.
Прогноз
Прогноз является переменным. Опасная для жизни осложнения включают аплазию костного мозга и лейкозную трансформацию, а иногда и вирусные инфекции. Около 1/3 пациентов имеют данные осложнение, некоторые из них могут быть успешно купированы трансплантацией костного мозга.
Врожденная амегакариоцитарная тромбоцитопения
Синонимы: врожденная амегакариоцитарная тромбоцитопеническая пурпура
Определение и общие сведения
Врожденная амегакариоцитарная тромбоцитопения является редким наследственным синдромом недостаточности костного мозга, характеризуется изолированным тяжелым уменьшением количества тромбоцитов и мегакариоцитов в первые годы жизни ребенка, которое позже развивается в недостаточность костного мозга с панцитопенией.
Точная распространенность неизвестна, менее чем 100 случаев описано в литературе. Кроме того, заболеваемость может быть недооценена из-за трудной диагностики заболевания. Наследование аутосомно-рецессивное.
Этиология и патогенез
Врожденная амегакариоцитарная тромбоцитопения происходит из-за мутаций в гене MPL (1p34), кодирующем рецептор (с-MPL) тромбопоэтина, экспрессирущий в плюрипотентных гемопоэтических стволовых клетках и в мегакариоцитах. Различные типы мутаций связаны с различными фенотипами патологии. Нонсенс-мутации приводят к полной потере функции рецептора тромбопоэтина — I тип, миссенс мутации ведут к остаточной функции рецептора — тип II. Случаи, без каких-либо дефектов в гене MPL рассматриваются как тип III.
Делеции 21q22 приводят к RUNX1 гаплонедостаточности — развивается вариант амегакариоцитарной тромбоцитопении с различными аномалиями: задержка роста, дефицит слуха, грыжи и трудности кормления.
Клинические проявления
Врожденная амегакариоцитарная тромбоцитопения проявляется с рождения, часто в первый день или, по крайней мере, в течение первого месяца жизни петехиями, пурпурой, желудочно-кишечными или внутричерепными кровоизлияниями, кровоизлияними в легких вследствие наличия изолированной тромбоцитопении и почти полным отсутствием мегакариоцитов в костном мозге. Тип I является тяжелой формой заболевания и характеризуется постоянно низким количеством тромбоцитов и раннем прогрессированием (как правило, в возрасте до 2-х лет) аплазии костного мозга, сопровождаемой панцитопенией. Тип II является более мягкой формой, которая проявляется транзиторным увеличением количества тромбоцитов, более 50×109/л, в течение первого года жизни или позже в возрасте 3-6 лет и не сопровождается развитием панцитопении.
Есть сообщения о случаях сопутствующих дефектов межпредсердной и межжелудочковой перегородок, гипоплазии головного мозга и мозжечка, а также замедления психомоторного развития.
Диагностика
Диагноз основывается на клинических признаках, тромбоцитопении (количество тромбоцитов ниже 50×109/л) с нормальным средним объемом тромбоцитов, значательном повышении уровня тромбопоэтина в сыворотке и отсутствии или малом количестве мегакариоцитов в аспирате костного мозга. Генетическое тестирование может подтвердить диагноз.
Дифференциальный диагноз
Дифференциальный диагноз проводят с идиопатической тромбоцитопенической пурпурой, на поздней стадии с апластической анемией, а также с анемией Фанкони, тромбоцитопенией с отсутствием лучевой кости и синдромом Вискота-Олдрича.
Лечение
Лечение поддерживающее, показаны переливания тромбоцитов. Трансплантация кроветворных стволовых клеток является единственной эффективной лечебной стратегией.
Прогноз
Прогноз неблагоприятный, трилинейная аплазия костного мозга происходит в течение первых лет жизни.
WT конечностно-кровяной синдром
Характеризуется гематологическими аномалиями (анемия Фанкони, лейкоз и лимфомы), которые часто обнаруживаются в детстве. Также присутствуют аномалии развития конечностей и кистей рук: расщепление или гипоплазия большого пальца, кожная синдактилия, дефекты локтевой и лучевой костей. Синдром был описан в нескольких семьях. Передача аутосомно-доминантная.
Синдром атаксии с панцитопенией
Синонимы: атаксия-панцитопения синдром, миелоцеребеллярный синдром
Синдром атаксии с панцитопенией характеризуется мозжечковой атаксией, различной гематологической цитопенией и предрасположенностью к недостаточности костного мозга и миелоидному лейкозу, иногда связан с моносомией 7 пары.
Вызвается миссенс мутацией гена SAMD9L. Наследование аутосомно-доминантное.
Источники (ссылки)[править]
https://www.orpha.net
Am J Hum Genet. 2016 Jun 2; 98(6): 1146–1158.
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Гидрокортизон
- Дексаметазон
- Метилпреднизолон
- Преднизолон
Источник

